HORMONES 2004, 3(3):171-183
DOI: 10.14310/horm.2002.——
Address correspondence and requests for reprints to:
Agathocles Tsatsoulis, MD, PhD, FRCP, Professor of Medicine/Endocrinology, Department of Medicine, Division of Endocrinology, University of Ioannina, 45110, Ioannina Greece, Tel: 26510-99625, Fax: 26510-46617,
e-mail: atsatsou@cc.uoi.gr
Received 11-03-04, Revised 06-05-04, Accepted 15-06-04
Abstract
The effects of estrogens on reproductive tissues and climacteric symptoms are unambiguous. However, their effects on other tissues and, in particular, the cardiovascular system remain controversial. In general, premenopausal women are protected from coronary heart disease (CHD) compared with aged-matched men but this `female protection’ appears to be lost after menopause, suggesting beneficial effects of female sex hormones on the cardiovascular system. This view has been supported by observational studies showing that estrogen replacement therapy (ERT) is associated with a 30% to 50% decrease in CHD risk in postmenopausal women. Nevertheless, randomized clinical trials in postmenopausal women, with or without pre-existing CHD, have found no benefits to combined hormone replacement therapy (HRT). A possible explanation for these apparently contradictory findings may relate to the divergent effects of estrogens depending on the state of the vascular endothelium. It has been suggested that estrogens may prevent the development and early progression of atherosclerosis by contributing to the maintenance of endothelial health but may have a neutral or negative effect on a dysfunctional endothelium or on advanced atheromatous lesions. Furthermore, it is possible that estrogens have adverse effects on other cardiovascular risk factors such as thrombosis and inflammation. It is also conceivable that a decline in vascular estrogen receptor expression with advancing age, through methylation of the estrogen receptor promoter, prevents postmenopausal women from deriving the beneficial cardiovascular effects of estrogens. In conclusion, estrogens possibly prevent the development of atherosclerosis through favourable effects on an intact endothelium, but once the vascular endothelium is damaged, the prothrombotic and possibly proinflammatory effects of estrogens are likely to predominate and prove harmful.
Key words: Estrogens, Hormone replacement therapy, Cardiovascular disease, Estrogen receptors
INTRODUCTION
Epidemiological data suggest that premenopausal women are largely protected from coronary heart disease (CHD) compared with men of similar age1. This phenomenon, referred to as `female advantage’, is gradually lost after menopause, so that by the sixth decade women have the same incidence of CHD as men. The protection of premenopausal women from CHD has been attributed to the cardioprotective effect of female sex hormones1. Estrogens are involved in many physiological processes which are known to be important for cardiovascular health in women and were until recently considered to protect women from cardiovascular disease (CVD). Extensive evidence from observational studies support this view, especially in regard to CHD2,3. However, randomized clinical trials in postmenopausal women, with or without CHD, have shown no benefits from combined hormone replacement therapy (HRT), casting doubt on the cardioprotective effect of estrogens on postmenopausal women4,5.
It has recently been suggested that the loss of `healthy vascular endothelium’ may prevent women from deriving cardioprotective benefits from endogenous or exogenous estrogens6. According to this hypothesis, the favourable antiatherogenic and other favourable vascular effects of estrogens are endothelium-dependent and receptor-mediated. Consequently, endothelial injury or decline in vascular estrogen receptor expression can diminish the cardiovascular benefits of the sex hormones. This concept may in part explain the disparity between the observational studies showing a cardioprotective effect of estrogens in healthy postmenopausal women and the recently published clinical trials on secondary and primary prevention of CHD in postmenopausal women with HRT which showed a different effect4,5.
In this review, we report on the biological effects of estrogens on the vascular system and in particular on endothelial function, and we analyze the divergent results from the observational studies and randomized clinical trials. It seems that the likely role of estrogens is to contribute to the maintenance of a healthy vascular endothelium but, in the presence of endothelial injury and the atheromatous vascular wall, the use of HRT could be harmful.
SOURCES OF ESTROGENS IN WOMEN
The naturally occurring estrogens 17β-estradiol (E2), estriol (E3) and estrone (E1) are C18 steroids derived from cholesterol through a series of enzymatic reactions in steroidogenic cells (Figure 1).

Figure 1. Sex steroid biosynthetic pathways. Enzymes: 1: cholesterol-20 and 22-desmolase, 2: 3β-hydroxysteroid deydrogenase, 3: 17α-hydroxylase, 4: 17- and 20-desmolase, 5: aromatase, 6: 17-hydroxysteroid deydrogenase.
In women of reproductive age, estrogens are produced mainly in the ovaries. Under the influence of luteinizing hormone (LH) the ovarian theca cells produce two androgenic steroids, androstendione and testosterone, which are aromatized in granulosa cells into E1 and E2, respectively, by the action of follicle stimulating hormone (FSH). Following ovulation, the luteinized cells produce progesterone during the luteal phase of the menstrual cycle. Progesterone has two roles: transformation of the endometrium after estrogen priming (luteominetic effect), and antiestrogenic effect limiting proliferation of the endometrium.
In premenopausal women E2 levels range from 10-80pg/ml during the follicular phase and reach the level of 600pg/ml at midcycle. After menopause the concentration of E2 falls to 5-30pg/ml. The main estrogen in menopausal women is E1, resulting from aromatization in adipose tissue of androstenedione which is produced by the adrenals.
In the circulation, estrogens bind mainly to sex hormone binding globulin (SHBG) produced in the liver and only a small amount of estrogens (2-3%) remains free7. Changes in SHBG levels may influence the tissue availability of free estrogens but also of free androgens, since the latter are also bound to SHBG. Although estrogens themselves increase SHBG levels, androgens and high insulin levels have the opposite effect. In menopause, the fall of estrogens in women is associated with reduced SHBG leading to decreased binding and thus to an increase in the level of free androgens. Estrogens fall to a greater extent and the androgen/estrogen ratio rises. The alteration in the balance between androgens and estrogens may contribute to changes in body composition and other symptoms associated with the menopause8.
In addition to endogenous estrogens, there are also exogenous sources of estrogen such as the oral contraceptives (OC) and hormone replacement therapy (HRT). The OCs consist of ethinyl-estradiol combined with a synthetic progestogen. For HRT, conjugated equine estrogens (CEE) or other oral or transdermal forms of synthetic estrogens are used in association with an orally administered progestogen. The progestogen is administered either cyclically or in a continuous combined regimen and is included in order to avoid the risk of endometrial hyperplasia9.
Selective estrogen receptor modulators (SERMs) belong to another category of chemical substances that have an estrogenic like activity with regard to vascular endothelium and bones, but an anti-estrogenic effect on the mammary gland and endometrium10. Tibolone is a new synthetic steroid with a progestogen-like structure that is converted to estrogenic and androgenic derivatives in different tissues and is also used as HRT11. Finally, phytoestrogens, derived from various plants, have an estrogen-like activity that may help in reducing climacteric and menopausal symptoms12.
ESTROGEN RECEPTORS AND MOLECULAR EFFECTS
Estrogens act in target tissues by binding to estrogen receptors that belong to the large family of nuclear ligand-activated transcription factors13. Once bound by estrogen, the estrogen receptor undergoes a conformational change, allowing the receptor to bind with high affinity to specific DNA-sequences or response elements (EREs) and modulate transcription of target genes14. Two types of estrogen receptors are known; estrogen receptor-α (ERα) and estrogen receptor-β (ERβ), encoded by different genes. Both receptor types are found in the cells of the cardiovascular system as well as in various other human tissues. ERβ is distributed differently from ERα within the tissues of the body and probably mediates different cell functions, but this is a field that needs further investigation.
Several studies have demonstrated the presence of functional estrogen-receptors in vascular endothelial cells as well as in smooth muscle cells15,16. In women with premature atherosclerosis, a decreased ER expression on atherosclerotic arteries has been observed17.
Regarding the role of ER in the regulation of endothelial function, a number of studies using selective ER-antagonists have taken place. The results of these studies have shown that 17β-estradiol enhances the production and activity of the enzyme nitric oxide synthase (eNOS), either via classical effects on gene transcription (genomic regulation), or via a non-genomic pathway18. The latter refers to a rapid stimulation of endothelial NO production in an ER-dependent manner18,19. There is strong evidence that eNOS is targeted to the endothelial plasma membrane, particularly to caveolae, which are specialized membrane domains20-22. A subpopulation of ERα is localized to endothelial-cell caveolae where they are coupled to eNOS in a functional complex that mediates non-genomic activation of eNOS. The rapid stimulation of eNOS by 17β-estradiol is due to an increase in intracellular calcium, mediated by tyrosine-kinase/MAP-kinase activation23,24.
Since NO is the main vasodilator and the best antioxidant substance, its decreased production causes endothelial dysfunction. Thus, a reduced estrogenic action, either as a result of decreased estrogen levels or reduced number of functional ERα in blood vessels and endothelium, probably constitutes a considerable cardiovascular risk factor as it may lead to premature endothelial dysfunction and therefore to atherosclerosis25.
VASCULAR EFFECTS OF ESTROGENS
The blood vessel wall consists of smooth muscle cells and a lining of endothelial cells, both of which have functional ERs as referred to above. Consequently, estrogens are believed to play a significant role with regard to vascular homeostasis. Their principal effect appears to be on the regulation of vascular tone. Generally, two types of factors modulate vascular tone: vaso-constrictive, such as endothelin-1, thromboxane, prostaglandin H2 and angiotensin, and vaso-relaxing factors. The predominant relaxing factor is NO, produced by the endothelial cells. Release of NO activates smooth muscle cell quanylate cyclase, which in turn causes an increase in cGMP production leading to vasodilation26. Other relaxing factors, such as prostacyclin and hyperpolarizing factor, act through cAMP and potassium channels. The NO release also has an anti-inflammatory and anti-atherogenic effect because it prevents platelet aggregation and suppresses smooth muscle cell proliferation27.
These two actions prevent endothelial dysfunction which is regarded as an early manifestation of atherosclerosis28,29.
The clinical evaluation of endothelial function is based on the observation of vasodilative ability after pharmacological or mechanical stimuli30. For this purpose three techniques have been adopted. The first two exploit the administration of acetylcholine intracoronary and intraarterially, which provides a dilatory effect and an increase in blood-flow, respectively. The third technique uses increased blood-flow shear (flow-mediated) as a mechanical means to estimate vasodilation through NO release30. The most commonly used technique is measurement of branchial artery diameter changes with high-frequency ultrasound after blood pressure cuff-induced hyperemia31. The use of nitroglycerine (TNT) or nitroprusside helps to estimate the non-endothelium dependent vasodilation.
There is evidence that estrogens contribute to the maintenance of “endothelial health”. In fact, the arterial endothelial NO seems to be the primary vascular target of estrogens32 which exert two types of effects with regard to endothelial function.
First, estrogens have long-term effects which are mediated at the level of gene transcription (genomic regulation). These effects are due to an increase in the expression and activity of eNOS, as explained above18,32,33. Furthermore, it has been observed that estrogens (particularly unopposed CEE) reduce plasma levels of endogenous assymetric dimethylarginine (ADMA)34, an inhibitor of eNOS35 and a cardiovascular risk factor36.
Clinical observations support these effects. Not only do estrogens cause vasodilation and increased blood-flow, but these responses also depend on plasma levels of estrogens. The observations that in young women endothelium-dependent vasodilation in branchial artery varies with the phase of the menstrual cycle, favours this conclusion37,38. Researchers have used blood pressure cuff-induced hyperemia to study endothelial function in premenopausal and postmenopausal women and found that there were greater vasodilatory responses in premenopausal women. When TNT or nitroprusside was used, the responses in pre- and postmenopausal women were quite similar39-41. Estrogen replacement therapy (ERT) given to postmenopausal women enhanced endothelium-dependent dilation in the branchial and coronary arteries42,43.
Similar results were observed in women with premature ovarian failure or ovariectomy after the use of estrogen, as well as in young women who used oral contraceptives41,44. It is of interest that inclusion of progesterone in postmenopausal HRT may blunt the effects of estrogens on endothelial NO production44.
In addition, estrogens exert short-term effects on the endothelium, causing rapid dilation by both endothelium-dependent and endothelium-independent pathways33. Thus, estrogens can cause rapid vasodilation (within minutes) when given intravenously or intraarterially in postmenopausal women45. These effects are probably mediated by a subpopulation of ERα localized to caveolae in endothelial cells and coupled to eNOS in a non genomic manner, as mentioned above20-22.
Estrogens also exert additional effects. These include stimulation of prostacycline, that causes vasodilation, and inhibition of endothelin-1-production in human vascular endothelial cells46. In addition, estrogens prevent apoptosis in human vascular endothelial cells and inhibit the migration and proliferation of smooth muscle cells in vitro47-49.
SYSTEMIC EFFECTS OF ESTROGENS
Effects on lipids and lipoproteins
Estrogens have beneficial effects on lipids and lipoproteins, but these effects depend on the type of estrogen used, the route of administration, the co-administration of progestogen and the type of progestogen50.
In general, oral estrogens reduce cholesterol accumulation in peripheral tissues and increase its biliary excretion51,52. In particular, estrogens decrease total cholesterol and low-density-lipoprotein (LDL) cholesterol by about 5-15%, due to an increase in the liver LDL-receptors, and enhancement of LDL-catabolism and clearance. Estrogens also reduce apolipoprotein (a) {Lp (a)} and increase high-density-lipoprotein (HDL) cholesterol by about 10% by decreasing HDL-receptors in liver cells. On the other hand, estrogens increase plasma triglycerides (TG) by 20-25%.
In addition to its effect on serum lipid profile, estradiol has an antioxidant capacity so that when 17β-estradiol is given to menopausal women, it reduces oxidation of LDL cholesterol and enhances endothelial NO bioactivity53.
Transdermal estrogens do not affect serum lipids and lipoproteins and this is probably related to the absence of a first-pass hepatic effect54. On the other hand, progestogens have no influence on LDL or Lp(a), but they appear to have an antiestrogenic activity on HDL and TRGs50.
Recent evidence suggests that remnant lipoprotein particles (RLPs) are the most atherogenic lipid particles. RLPs were associated with endothelial dysfunction and atherosclerosis and were identified as an independent risk factor for cardiovascular disease in women55. A recent randomized study has shown that HRT has beneficial effects onRLPs, with no influence on triglycerides56.
Effects of estrogens on hemostasis
Hemostasis is a natural mechanism of the human body aimed at halting bleeding irrespective of its cause. For this purpose a number of coagulant factors play an important role. On the other hand, there are regulatory factors, i.e. anticoagulant factors and the fibrinolytic system, with opposite effects, so that hemostasis does not lead to vessel obstruction. An outline of this hemostatic balance is given in Figure 2.
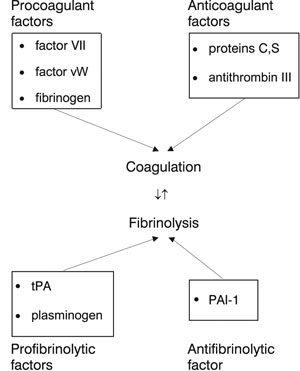
Figure 2. Hemostatic balance. Abbreviations: Factor vW:Factor von Willebrant, tPA:tissue plasminogen activator, PAI-1: Plasminogen activator inhibitor type 1.
Estrogens appear to interfere in the hemostatic balance as they regulate, through estrogen receptors, the expression of genes for a number of coagulant and fibrinolytic proteins57. This regulation is probably under genetic control because the hemostatic system of some women appears to be more sensitive than that of other women58.
It has been reported that in postmenopausal women the levels of factor VII, factor vW and fibrinogen are elevated59. These factors are important risk markers for ischemic heart disease. On the other hand, the use of HRT in postmenopausal women decreases the level of fibrinogen, but also decreases protein S and antithrombin III and enhances activity of factor VII60.
In addition, increased levels of PAI-1 have been found in postmenopausal women, leading to fibrinolysis inactivation that is associated with atherosclerosis61. Even small doses of ERT reduce PAI-1 and activate the fibrinolytic system. The combination with progestogens maintain the same beneficial effects, although transdermal estrogens do not influence PAI-162,63. The activation of fibrinolysis does not appear to be dose-dependent, unlike the coagulatory activity61.
Effects of estrogens on inflammatory markers
Atherogenesis and the ensuing plaque disruption are the result of an inflammatory process that takes place in the vessel wall. C-reactive protein (CRP) is the most important marker of inflammation and the strongest independent predictor of cardiovascular events in apparently healthy women64,65.
There is evidence that the level of CRP rises in postmenopausal women using oral estrogens66,67, although the latter reduce other inflammatory markers such as E-selectin VCAM-1, ICAM-1 and soluble thrombomodulin68. This indicates that the rise in CRP levels is not the result of an increased inflammatory response but has to do with a metabolic liver-activation. By contrast, studies in animals where estrogens were given subcutaneously or intravenously failed to demonstrate such pro-inflammatory effects69. It appears that the route of administration of estrogen may be important with regard to its adverse effects on inflammatory markers65.
Thus, the use of HRT in women with pre-existing coronary heart disease may become problematic70. HRT reduces serum levels of the cell adhesion molecules that may inhibit attachment of white blood cells to the vessel wall, but this therapy also causes a rise in serum levels of matrix metalloproteinase-9 (MMP-9). In addition, HRT decreases plasma levels of PAI-1 leading to plasmin activation, which may also activate metalloproteinases. These events in women with atheromatous plaques could result in digestion of the matrix proteins of the fibrous cap and therefore provoke thrombosis71,72.
HORMONE REPLACEMENT THERAPY AND CARDIOVASCULAR DISEASE: CLINICAL STUDIES
Premenopausal women develop CVD less frequently than men of similar age. However, the protection that women appear to have during their reproductive age fades after menopause when the rate of coronary heart events tends to be equal to that of men73. Among the measures that have been taken over the last few decades to reduce the risk of CVD are lifestyle changes and pharmacologic interventions. HRT was among the pharmacologic interventions and was thought to have beneficial effects in postmenopausal women74,75.
Observational studies
A large number of observational studies have suggested that the use of exogenous estrogens might be cardioprotective. In a review of population-based, case-control, cross-sectional and prospective studies of estrogen therapy (in the form of CEE), the relative risk for CHD was reduced by approximately 50%76. In current hormone users there were reductions in CHD-risk which ranged from 35% to 50%, compared to non-users75,77. In the Nurses’ Health Study, in which 70,533 postmenopausal women were followed-up for 20 years, the relative risk of major coronary events was 0.61(CI: 0.52-0.71) forcurrent use of estrogens, adjusted for age and the common cardiovascular risk factors3. In a recent study, the progress of coronary atherosclerosis was estimated by measurement of coronary artery calcium (CAC) with electron beam computed tomography (EBT)78. The participants of this study were 2,213 healthy postmenopausal women with a mean age of
59 years; half of the women were current users of HRT. The results showed that current HRT use decreased the possibility of high calcium score by 50%.Therefore, these results indicate that HRT use is associated with less coronary artery atherosclerosis as estimated by CAC, since high calcium scores are significant predictors of cardiovascular events in most large observational studies79,80. In addition, the study showed that the longer women received HRT, the greater was the fall in the calcium score (dose-response relationship).
In general, observational studies promoted the belief that HRT plays a cardioprotective role. However, in a recent observational study in women with established coronary heart disease, the use of HRT was harmful. There was an increase in the cardiovascular events in women who started HRT after a myocardial infarction81.
With regard to the observational studies there are two important issues worthy of discussion. First there is the problem of bias. In these studies comparisons were made between women who have elected to take HRT and women who either elected not to take HRT or who themselves had not considered taking it (selection bias)82. These two groups of women may have had different lifestyles and educational or socioeconomic status. Women who used HRT were likely to have also taken more preventive health measures compared to women not on HRT, therefore, they were also likely to be at lower risk for CHD regardless of HRT use83. However, in the Nurses’ Health Cohort Study, in which users and non-users had the same social status and educational level, hormone use also appeared to be protective3.
The second issue concerns the type of hormone regimen used, as women in the observational studies used mainly unopposed CEE. On the other hand, it is known that progestogens may oppose some of the estrogen effects on the cardiovascular system84. However, a limited number of observational studies with combined HRT suggest that the effect of progestogen may not be substantial, although the type of progestogen used may be relevant.
As the role of HRT in menopause remained inconclusive, the need for clinical trials in order to clarify the potential role of HRT on cardiovascular disease in postmenopausal women became apparent.
Randomized clinical trials
The Heart and Estrogen/Progestin Replacement Study (HERS) was the first large clinical trial to test the use of HRT as secondary prevention in women with established coronary heart disease4. HERS was a double-blind, placebo controlled, randomized study of 2,763 postmenopausal American women, of mean age 66.7 years, who received combined continuous oral HRT {0.625mg CEE plus 2.5mg MPA (medroxyprogesterone-acetate) daily} or placebo. The trial was ended prematurely after 4.1 years because there was an increase in the coronary and thromboembolic events in women who used HRT during the first year, although the serum lipid profile had improved. By the fourth year of study, cardiovascular events were fewer in the HRT-users than in the placebo group. However, recently published data from an extension of the HERS study to 6 years (HERS II) have indicated that the trend towards reduction in cardiovascular events did not continue85. Because of an increase in the risk of venous thrombosis among HRT-users, it was suggested that HRT is not beneficial in women with established cardiovascular disease who already have atherosclerotic plaques prone to rupture.
There was some criticism of the HERS trial results86. The first criticism was the older age of women participating in the study. It was then suggested that lower doses of estrogens might be more beneficial with regard to the progress of atherosclerosis. Another issue was the fact that in HERS a specific combination of hormones was used (CEE+MPA). Whether the results would be the same if other combinations of estrogen+progestogen were used remained unanswered. The answer to this came from another study, the Papworth Hormone Replacement Therapy Atherosclerosis Study. In this study postmenopausal women used transdermal 17β-estradiol alone or in combination with norethisterone. Again, this regimen had no benefit in reducing CHD events in women with pre-existing CVD87.
The second clinical trial was the Estrogen Replacement and Atherosclerosis (ERA), a randomized trial in postmenopausal women who had angiographically defined CHD at baseline in order to evaluate coronary artery changes after the use of HRT88. Again, neither CEE alone nor in combination with MPA were effective in slowing down the progress of atherosclerosis in coronary arteries, although beneficial effects on lipid profile were noted.
It was concluded from these studies that HRT should not be used for secondary prevention of CHD. Subsequently, the role of HRT for primary prevention of CHD was addressed.
To this end, a large clinical trial, the Women’s Health Initiative (WHI), was designed5,89. It was a randomized, double-blind, placebo-controlled, clinical trial of 16,608 postmenopausal women, aged 50 to 71 years (mean age 63.7 years), who received either continuous oral combined CEE (0.625mg) + MPA (2.5mg) or placebo daily. Another group of 10,739 postmenopausal women without uterus, as a result of hysterectomy, used CEE alone or placebo. This part of the study is still running. However, the first part was stopped after 5.2 years of follow-up because the risks exceeded the benefits. There was an increased risk of invasive breast cancer (8 cases/10,000 women), which rose the longer the women continued the therapy. The risk of non-fatal myocardial infarctions and coronary death also increased (7 cases/10,000 women) especially during the first year of the study. The risk of venous thromboembolism was doubled (18/10,000 women). Finally, there was an excess risk of stroke (8/10,000) throughout the trial. These results disappointed the optimists among the scientists who believed that HRT could prevent CVD in healthy postmenopausal women. On the other hand, some benefits from the use of HRT were identified. The incidence of hip fractures was reduced (8/10,000), as was the incidence of colorectal cancer (6/10,000). There was no overall benefit, however, over the 5.2 years of the trial.
The Estrogen in the Prevention of Atherosclerosis Trial (EPAT) followed up postmenopausal women without preexisting CHD in order to estimate the effect of the oral use of 1mg 17β-estradiol in the progress of carotid atherosclerosis90. Women in whom serum LDL was above 160mg/dl received antilipid therapy. The results showed that these women had no alterations in the carotid artery ultrasound after two years of oral therapy. In contrast, in women with low serum LDL-levels, the use of 17β-estradiol slowed the progression of carotid intima-media thickness in comparison to placebo. Overall, this trial showed a beneficial effect of estrogen use in healthy postmenopausal women with regard to atherosclerosis of the carotid arteries. On the other hand, another clinical trial, the WELL-HART (Women’s Estrogen/Progestin Lipid Lowering Hormone Atherosclerosis Regression Trial) in postmenopausal women with established coronary artery atherosclerosis, showed that the use of 17β-estradiol alone or in combination with MPA had no influence on the progression of atherosclerosis evaluated by coronary angiography, compared to placebo91.
Surprisingly, therefore, the results from randomized clinical trials of HRT use in postmenopausal women for primary or secondary prevention of CHD do not support the observational data on the cardioprotective role of estrogens. Several possible explanations for the divergent data on postmenopausal HRT have been suggested.
The complexity of sex steroid effects
The main estrogen in the HRT used in the clinical trials is CEE which is usually prescribed in the U.S.A. On the other hand, 17β-estradiol is more commonly used in Europe, but there have been no observational data or large clinical trials for primary prevention with 17β-estradiol86.
Another possible explanation might be the high dose of estrogens used. There is evidence that lower doses of estrogen have beneficial effects on lipoproteins and coagulant factors. Furthermore, older women, such as the participants in the two large clinical trials, may be more susceptible to inflammatory and thrombotic effects of higher doses of estrogen92.
On the other hand, it is possible that the use of progestogen in combination with estrogen may mask estrogen’s cardioprotective role86. This will probably be clarified at the end of the second part of the WHI study in which 10,739 women without uterus received CEE alone or placebo.
In addition, oral estrogens increase CRP66,67 and induce expression of matrix metalloproteinases within the vessel wall. The latter can digest and weaken fibrous caps of vulnerable plaques, leading to thrombosis71,72.
Another criticism of the WHI trial is the fact that the women participating were older, so that they were likely to already have existing atherosclerotic changes. Since atherosclerosis down-regulates estrogen receptors, the transcription of multiple genes, some of which are responsible for the potential protective ef
fects of estrogen, is reduced and this fact cannot be excluded86.
The state of the vascular endothelium
Experimental and clinical evidence suggests that the effects of estrogens are dependent on the integrity and functional status of the vascular endothelium and the presence or absence of atherosclerotic changes in the vascular wall. This `healthy endothelium’ hypothesis may in part explain the unfavourable findings of the HERS and WHI trials and guide future strategies of the use of HRT6.
According to this hypothesis, endothelial injury or decline in vascular estrogen receptor expression can diminish the antiatherogenic properties of estrogens. Indeed, experimental studies have examined the effects of endothelial damage induced by balloon catheter injury in rabbits and how estrogens affect progression of atherosclerosis. It was found that the antiatherogenic effects of estrogens were present, absent or reversed depending on the state of the arterial endothelium93.
Studies in oophorectomized cynomolgous monkeys also support this hypothesis. Thus, in monkeys assigned to CEE (alone or in combination with MPA) beginning two years (approximately six human years) after oophorectomy and well after atherosclerosis was established, HRT had no effect on the extent of coronary artery plaque. However, when HRT was given to monkeys immediately after the oophorectomy, during the early stages of atherosclerosis, there was a 50% reduction in the extent of plaque formation94. In this regard, in the Nurses’ Health Study women ranged in age from 30 to 55 years at enrollment and almost 80% began estrogens within two years after menopause3. In contrast, the mean age of the participants in the HERS and in the WHI trials were 67 and 63 years, respectively, and thus on average 10 years postmenopausal at the time of enrollment, an age at which atherosclerosis had already set in.
Furthermore, studies in humans have shown that non-diseased coronary artery vessels dilate in response to the administration of estrogens, whereas diseased vessels do not respond95. The lack of estrogen receptor expression in the presence of atherosclerosis may result in a decreased ability of vascular tissue to respond to estrogens17. In this regard, down-regulation of ERs, may have an impact on the effectiveness of estrogen. It is well established that an important mechanism for down-regulation of gene-expression is methylation of a cytosine and guanine rich area in the promoter-region of the ERα-gene, called CpG-island. Methylation leads to permanent inactivation of gene-transcription in multiple systems96.
It has been shown that age and perhaps vascular injury are associated with de novo methylation of the ERα-gene promoter in smooth muscle cells. These cells lose some of the normal growth control and, under certain conditions, proliferate abnormally and may contribute to the pathogenesis of the atheromatous plaque. Another scenario is the possibility that ERα-gene-methylation is the result of atherosclerosis and hyperproliferation97.
It is tempting to speculate that in women with advanced atherosclerosis, ERα-gene methylation in diseased vessels may contribute to the lack of benefit after the use of HRT observed in the clinical trials with older postmenopausal women97. According to recent research evidence, methylation may possibly be reversed pharmacologically by the use of inhibitors of the DNA-Methyltransferase enzyme98.
ALTERNATIVE THERAPIES TO HRT
Since the use of conventional HRT regimens has already been shown to have disadvantages, scientists have turned their interest to alternative therapies in order to minimize the side effects of estrogen and explore their beneficial effects.
The first category of these new therapies consists of the Selective Estrogen Receptor Modulators [SERMs], which are non-steroidal estrogen compounds with an estrogen like effect (on lipoproteins and bones) as well as an estrogenic antagonist effect on the mammary gland and endometrium7. Raloxifene reduces the level of LDL-cholesterol and fibrinogen in plasma, but has no effect on HDL levels99. There have been various studies with divergent results. In ovariectomized cholesterol-fed rabbits, a reduction in aortic atherosclerosis was reported with the use of raloxifene but to a lesser extent than that with the use of 17β-estradiol100. Studies in monkeys showed no beneficial effect of raloxifene99. In the MORE trial (Multiple Outcomes of Raloxifene Evaluation Trial), with more than 7,000 postmenopausal women participating, raloxifene was shown to have no benefits compared with placebo in regard to CHD and cardiovascular events, but this was not the primary aim of the study. However, in women with increased risk of cardiovascular disease there were fewer events compared with placebo101. Another study, the RUTH trial (Raloxifene Use for The Heart Study), is currently testing the impact of raloxifene on cardiovascular end-points in postmenopausal women. The results of this trial will provide further information on the role of SERMs in CVD.
Another category, with tibolone as first representative, is the selectivetissueestrogenicactivity regulators(STEAR). STEAR have a progestogen-like structure that is converted to estrogenic and androgenic derivatives in-vivo. Tibolone is effective in the control of climacteric symptoms (due to its 3-OH metabolites) and in maintaining bone density, without stimulating tissues such as the endometrium and mammary gland102,103. Thus tibolone is different from estrogens and SERMs as it reduces menopausal symptoms and the risk of thromboembolism (it promotes fibrinolysis rather than thrombosis). Tibolone also decreases not only triglycerides and Lp (a) levels but also HDL-cholesterol104,105. However, there have been no clinical trials on the effect of tibolone on the primary prevention of cardiovascular disease in postmenopausal women.
Finally, within the spectrum of alternative therapies to HRT are the phytoestrogens, a group of natural compounds derived from herbs with estrogen agonist and antagonist properties106. The strongest estrogenic activity has been observed in soy in which the removal of a glucone-group significantly raises its bioactivity106-110. High estrogen activity was also discovered in extract of fo-ti, not previously reported106. Soy phytoestrogens have shown beneficial effects on endothelium-dependent vasodilation in the development of atherosclerosis in non-human primates108,111. Some studies in postmenopausal women, though not all, have shown beneficial effects on lipid profile and in endothelial function112. Clinical end-point data are not available, however, so that recommendations regarding the use of phytoestrogens for the prevention of CVD cannot be formulated as yet.
CONCLUSIONS AND FUTURE DIRECTIONS
Based on the favourable effects of estrogens on various tissues and functions, including the cardiovascular system, clinicians hoped that the use of estrogen alone or in combination with progestogen as replacement therapy in postmenopausal women would offer, among other benefits, cardioprotection. Unfortunately, clinical trials have not confirmed the cardioprotective role of HRT. It appears that in postmenopausal women with established coronary heart disease, HRT use might in fact be harmful because it increases cardiovascular events and the risk of venous thrombosis. Possible explanations for the divergent data between the observational studies and clinical trials include the type of estrogen and the high doses used, the possible antagonistic effects of progestogen, the age of the women who participated in the clinical trials and the state of the endothelium and stage of atherosclerosis at the time of intervention.
The early danger for both coronary heart disease and stroke observed in both the HERS and WHI trials may be a thrombotic effect, reflecting a synergistic effect between one of the relatively common hypercoaculable states and hormone therapy113. Another possible explanation is the pro-inflammatory effect of oral estrogens, with rupture of vulnerable plaques86.
Although clinical trials showed not only lack of benefits but also an adverse effect on the cardiovascular system after the use of HRT, one cannot ignore studies showing the beneficial effect of estrogens in the early stages of atherogenesis during the menopausal transition and the early years after menopause. In the ensuing years new strategies should be adopted in clinical trials. Thus, the use of lower doses of estrogen or combinations with different progestogen, novel estrogen agonists including vascular SERMs, a combination of SERMs or phytoestrogens with low-dose estrogens and better timing of intervention are possibly some of the future directions for Hormone Replacement Therapy.
On the other hand, it is important to emphasize behaviour changes and, where appropriate, lipid and blood pressure medication, which have been proved to reduce CHD-risk in postmenopausal women, should be considered86. For the time being, however, HRT is suspended for the primary and secondary prevention of CHD in postmenopausal women.
REFERENCES
1. Sullivan JM, Fowlkes LP, 1996 The clinical aspects of estrogens and the cardiovascular system. Obstet Gynecol 87: Suppl 2: 36-43.
2. Stampfer MJ, Colditz GA, Willett WC, et al, 1991 Postmenopausal estrogen therapy and cardiovascular disease; ten year follow-up from the Nurses’ Health Study. N Engl J Med 325: 756-762.
3. Grodstein F, Manson JE, Colditz GA, et al, 2000 A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 133: 933-941.
4. Hulley S, Grady D, Bush T, et al, 1998 Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women: Heart and EstrogenProgestin Replacement Study (HERS) Research group. JAMA 280: 605-613.
5. Manson JM, Hsia J, Johnson KC, et al, 2003 Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med 349: 523-534.
6. Koh KK, 2003 Can a healthy endothelium influence the cardiovascular effects of hormone replacement therapy? Int J Cardiol 87: 1-8.
7. Selby C, 1990 Sex Hormone Binding Globulin; origin, function and clinical significance. Ann Clin Biochem 27: 532-554.
8. Carr MC, 2003 The emergence of the metabolic syndrome with the menopause. J Clin Endocrinol Metab 88: 2404-2411.
9. Yen SSC, Jaffe RB, Barbieri RL 1999 Reproductive Endocrinology, Philadelphia Saunders (eds); pp, 110-133, 301-319, 751-784.
10. Bryant HV, Dere WH, 1998 Selective Estrogen Receptor Modulators; an alternative to Hormone Replacement Therapy. Proc Soc Exp Biol Med 217: 45-52.
11. Crook D 2001 Cardiovascular risk assessment for postmenopausal Hormone Replacement Therapy such as Tibolone [Livial]. HRT and cardiovascular disease. In: Genazzani AR (ed) New York, Parthenon; pp, 165-172.
12. Knight DC, Eden JA, 1995 Phytoestrogen; a short review. Maturitas 22: 167-175.
13. Weigel NL, 1996 Steroid hormone receptors and their regulation by phosphorylation. Biochem J 319: 657-667.
14. Murdoch FE, Gorski J, 1991 The role of ligand in estrogen receptor regulation of gene expression. Mol Cell Endocrinol 78: C103-108.
15. Kim-Schultze S, McGowan KA, Hubchak SC, et al, 1996 Expression of an estrogen receptor by human coronary artery and umbilical vein endothelial cells. Circulation 94: 1402-1407.
16. Venkov CD, Rankin AB, Vaughan DE, 1996 Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation 94: 727-733.
17. Losordo DW, Kearney M, Kim EA, Jekanowski J, Isner JM, 1994 Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation 89: 1501-1510.
18. Caulin-Glaser T, Garcia-Cardena G, Sarrel P, Sessa WC, Berder JR, 1997 17-β estradiol regulation of human endothelial cell basal nitric oxide release independent of cytosolic Ca mobilization. Circ Res 81: 885-892.
19. Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW, 1997 Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol 273: L116-126.
20. Schaul PW, Smart EJ, Robinson LJ, et al, 1996 Acylation targets endothelial nitric oxide synthase to plasmalemmal caveolae. J Biol Biochem 271: 6518-6522.
21. Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC, 1996 Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA 93: 6448-6453.
22. Schaul PW, Anderson RGW, 1998 Role of plasmalemmal caveolae in signal transduction. Am J Physiol 275: L843-851.
23. Fleming I, Fissthaler B, Busse R, 1995 Calcium signaling in endothelial cells involves activation of tyrosine kinases and leads to activation of mitogen-activated protein kinases. Circ Res 76: 522-529.
24. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Schaul PW, 1999 Estrogen receptor α mediates the non genomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest 103: 401-406.
25. Rubanyi GM, Kauser K, Johns A, 2002 Role of estrogen receptors in the vascular system. Vascular Pharmacology 38: 81-88.
26. Rubanyi GM, 1993 The role of endothelium in cardiovascular hemostasis and diseases. J Cardiovasc Pharmacol 22: Suppl 4: 1-14.
27. Post MS, Verhoeven MO, Van der Mooren MJ, Kenemans P, Stehouwer CDA, Teerlink T, 2003 The effect of HRT on plasma levels of the cardiovascular risk factor ADMA; a randomized placebo-controlled 12-week study in healthy early postmenopausal women. J Clin Endocrinol Metabol 88: 4221-4226.
28. Liao JK 1999 Endothelial nitric oxide and vascular inflammation. In: Panza JA, Cannon Roi (eds) Endothelium nitric oxide and atherosclerosis. Armonk NY, Futura; pp, 119-132.
29. Quyyumi AA, 1998 Endothelial function in health and disease; new insights into the genesis of cardiovascular disease. Am J Med 105: 325-395.
30. Nabel EL, Selwyn AP, Ganz P, 1990 Large coronary arteries in humans are responsive to changing blood flow: an endothelium dependent mechanism that fails in patients with atherosclerosis. J Am Coll Cardiol 16: 349-356.
31. Celermajer DS, Sorensen KE, Gooch VM, et al, 1992 Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 340: 1111-1115.
32. Kleinhart H, Wallerath T, Euchenhofer C, Ihrig-Biedert I, Li H, Fostermann V, 1998 Estrogen increase transcription of the human endothelial NO-synthase gene; analysis of the transcription factors involved. Hypertension 32: 588-592.
33. Mendelsohn ME, 2000 Mechanisms of estrogen action in the cardiovascular system. J Steroid Biochem Mol Biol 74: 337-343.
34. Teerlink T, Neele SJM, de Jong S, Metelenbos JC, Stehouwer CDA, 2003 Estrogen replacement therapy lowers plasma levels of ADMA in healthy post menopausal women. Clin Sci 105: 67-71.
35. Vallance D, Leone A, Calver A, Collier J, Moncada S, 1992 Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339: 572-575.
36. Valkonen VP, Paiva H, Salonen JT, et al, 2001 Risk of acute coronary events and serum concentration of ADMA. Lancet 358: 2127-2128.
37. Kawano H, Motogama T, Kugiyama K, et al, 1996 Menstrual cyclic variation of endothelium-dependent vasodilation of the branchial artery; possible role of estrogen and nitric oxide. Proc. Assoc. Am Physicians 108: 473-480.
38. English JL, Jacobs LO, Green G, Andrews TC, 1998 Effect of the menstrual cycle on endothelium-dependent vasodilation of the branchial artery in normal young women. Am J Cardiol 82: 256-258.
39. Majmudar NG, Robson SC, Ford GA, 2000 Effects of menopause, gender and estrogen replacement therapy on vascular nitric oxide activity. J Clin Endocrinol Metab 85: 1577-1583.
40. Pinto S, Virdis A, Chiadoni L, et al, 1997 Endogenous estrogen and acetylcholine-induced vasodilation in normotensive women. Hypertension 29: 268-273.
41. Kravariti M, Papanikolaou E, Kazakos N, et al 2003 Endothelial dysfunction in women with premature ovarian failure. Sixth European Congress of Endocrinology, Lyon; p, 744.
42. Lieberman EH, Gerhard MD, Uehata A, et al, 1994 Estrogen improves endothelium-dependent, flow-mediated vasodilation in postmenopausal women. Ann Intern Med 121: 936-941.
43. Best PI, Berger PB, Miller VM, Lerman A, 1998 The effect of estrogen replacement therapy on plasma nitric oxide and endotheline-1 levels in postmenopausal women. Ann Intern Med 128: 285-288.
44. Sorensen KE, Doryp I, Hermann AP, Mosekilde L, 1998 Combined hormone replacement therapy does not protect women against the age-related decline in endothelium-dependent vasomotor function. Circulation 97: 1234-1238.
45. Gilligan DM, Badar DM, Panza JA, Quyyumi AA, Cannon RO III, 1994 Acute vascular effects of estrogen in postmenopausal women. Circulation 90: 786-791.
46. Mikkola T, Turunen P, Avela K, et al, 1995 17β-estradiol stimulates prostacyclin but not endotheline-1 production in human vascular endothelial cells. J Clin Endocrinol Metab 80: 1832-1836.
47. Spyridopoulos I, Sullivan AB, Kearney M, Isner JM, Losordo DW, 1997 Estrogen receptor-mediated inhibition of human endothelial cell apoptosis; estradiol as a survival factor. Circulation 95: 1505-1514.
48. Kolodgie FD, Jacob A, Wilson PS, et al, 1996 Estradiol attenuates directed migration of vascular smooth muscle cells in vitro. Am J Pathol 148: 969-976.
49. Caulin-Glaser T, Watson CA, Pardi R, Bender JR, 1996 Effect of 17β-estradiol on cytokine-induced endothelial cell adhesion molecule expression. J Clin Invest 98: 36-42.
50. Godsland IF, 2001 Effects of postmenopausal HRT on lipid, lipoprotein and apolipoprotein (a) concentrations; analysis of studies published from 1974-2000. Fertil Steril 75: 898-915.
51. Kovanen PT, Brown MS, Goldstein JL, 1979 Increased binding of LDL to liver membranes from rats treated with 17β-ethinyl estradiol. J Biol Chem 254: 11367-11373.
52. Tikkanen MJ, Nikkia EA, Kuusi T, Sipinen S, 1982 HDL-2 and hepatic lipase; reciprocal changes produced by estrogen and norgestrel. J Clin Endocrinol Metab 54: 1113-1117.
53. Sack NM, Rader DJ, O’Connor R, 1994 Estrogen and inhibition of oxidation of LDL in postmenopausal women. Lancet 343: 269-270.
54. Crook D, Stevenson JC, 1996 Transdermal hormone replacement therapy, serum lipids and lipoproteins. Br J Clin Pract 86: 17-21.
55. McNamara JR, Schah PK, Nakajima K, et al, 2001 Remnant-like particle [RLPs] cholesterole is an independent cardiovascular risk factor in women; results from the Framingham heart study. Atherosclerosis 154: 229-236.
56. Ossewaarde ME, Dallinga-Thie GM, Bots ML, et al, 2003 Treatment with HRT lowers RLPs in healthy postmenopusal women; results from a randomized trial. Eur J Clin Invest 33: 376-382.
57. Mendelsohn ME, Karas RH, 1994 Estrogen and the blood vessel wall. Curr Opin Cardiol 9: 619-626
58. Rosendaal FR, Helmerhorst FM, Vandenbrouke JP, 2002 Female hormone and thrombosis. Arterioscler Thromb Vasc Biol 22: 201-210.
59. Meilahn EN, Kuller LH, Mathews KA, Kiss JE, 1992 Hemostatic factors according to menopausal status and use of hormone replacement therapy. Ann Epidemiol 2: 445-455.
60. Nabulsi AA, Folsom AR, White A, et al, 1993 Association of HRT with various cardiovascular risk factors in postmenopausal women. N Engl J Med 328: 1069-1075.
61. Winkler UH, 1992 Menopause, HRT and cardiovascular disease. A review of haemostaseological findings. Fibrinolysis 6: Suppl 3: 5-10.
62. Koh KK, Mincemoyer R, Bui MN, et al, 1997 Effects of HRT on fibrinolysis in postmenopausal women. N Engl J Med 336: 683-690.
63. Kroon UB, Silfverstolpe G, Tengborn I, 1994 The effects of transdermal estradiol and oral conjugated estrogen on hemostasis variables. Thromb Haemost 71: 420-423.
64. Ross R, 1999 Atherosclerosis; an inflammatory disease. N Engl J Med 340: 115-126.
65. Ridker PM, Hennekens CH, Buring JE, et al, 2000 C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342: 836-843.
66. Cushman M, 2002 Effects of hormone replacement therapy and selective estrogen receptor modulators [SERMs] on markers of inflammation and coagulation. Am J Cardiol 90: Suppl: 7-10.
67. Cushman M, Leqault C, Barret-Connor E, et al, 1999 Effect of postmenopausal hormones on inflammation-sensitive proteins; the Postmenopausal Estrogen/Progestin Interventions [PEPI] Study. Circulation 100: 717-722.
68. Silvestri A, Gebara O, Vitale C, et al, 2003 Increased levels of CRP after oral HRT may not be related to an increased inflammatory response. Circulation 107: 3165-3169.
69. Nunomura W, Takakuwa Y, Higashi T, 1994 Changes in serum concentration and m-RNA level of rat CRP. Bioch Bioph Acta 1227: 74-78.
70. Zanger D, Yang BK, Addams J, et al, 2000 Divergent effects of hormone therapy on serum markers of inflammation in postmenopausal women with coronary artery disease on appropriate medical management. J Am Coll Cardiol 36: 1797-1802.
71. Wingrove CS, Garr E, Godsland IF, Stevenson JC, 1998 17β-estradiol enhances release of matrix metalloproteinase-2 from human vascular smooth muscle cells. Biochem Biophys Acta 1406: 169-174.
72. Galis ZS, Khatri JJ, 2002 Matrix metalloproteinases in vascular remodeling and atherogenesis; the good, the bad and the ugly. Circ Res 90: 251-262.
73. Colditz GA, Willette WC, Stampfer MJ, Rosner B, Speizer FE, Hennekens CH, 1987 Menopause and the risk of coronary heart disease in women. N Engl J Med 316: 1105-1110.
74. Mosca L, 2000 The role of HRT in the prevention of postmenopausal heart disease. Arch Intern Med 160: 2263-2272.
75. Grodstein F, Stampfer M, 1995 Epidemiology of coronary heart disease and estrogen replacement in postmenopausal women. Progress Cardiol 38: 199-210.
76. Stampfer MJ, Colditz GA, 1999 Estrogen replacement therapy and coronary heart disease; a quantitative assessment of the epidemiologic evidence. Prev Med 20: 47-63.
77. Rackley CE, 1995 Estrogen and coronary artery disease in postmenopausal women. Am J Med 99: 117-118.
78. Akhrass F, Evans AT, Wang Y, et al, 2003 HRT is associated with less coronary atherosclerosis in postmenopausal women. J Clin Endocrinol Metab 88: 5611-5614.
79. Raggi P, Callister TQ, Cooil B, et al, 2000 Identification of patients at increased risk of first unheralded acute myocardial infarction by electron beam computed tomography. Circulation 101: 850-855.
80. Arad V, Sparado LA, Goodman K, et al, 1996 Predictive value of electron beam computed tomography of the coronary arteries; 19-month follow-up of 1173 asymptomatic subjects. Circulation 93: 1951-1953.
81. Alexander KP, Newby LK, Hellkamp AS, et al, 2001 Initiation of HRT after acute myocardial infarction is associated with more cardiac events during follow-up. J Am Coll Cardiol 38: 1-7.
82. Hemminki E, Sihvo S, 1993 A review of postmenopausal hormone therapy recommendations; potential for selection bias. Obstet Gynecol 82: 1021-1028.
83. Barret-Connor E, 1991 Postmenopausal estrogen and prevention bias. Ann Intern Med 115: 455-456.
84. Adams MR, Register TC, Golden PC, Wagner ID, Williams JK, 1997 Medroxy-progesterone antagonizes inhibitory effects of CEE on coronary artery atherosclerosis. Arterioscler Thromb Vasc Biol 17: 217-221.
85. Grady D, Herrington D, Bittner V, et al, 2002 Cardiovascular disease outcomes during 6.8 years of hormone therapy. HERS follow-up [HERS II]. JAMA 288: 49-57.
86. Elizabeth Barret-Connor, 2003 An epidemiologist looks at hormones and heart disease in women. J Clin Endocrinol Metab 88: 4031-4042.
87. Clarke SC, Kelleher T, Lloyd _Jones H, Slack M, Schofiel PM, 2002 A study of hormone replacement therapy in postmenopausal women with ischemic heart disease; the Papworth HRT atherosclerosis study. Br J Obstet Gynecol 109: 1056-1062.
88. Herrington DM, Reboussin DM, Brosnihan KB, et al, 2000 Effects of estrogen replacement on the progression of coronary artery atherosclerosis. N Engl J Med 343: 522-529.
89. Writing group for the Women’s Health Initiative Investigators, 2002 Risks and benefits of estrogen plus progestin in healthy postmenopausal women; principal results from the WHI randomized controlled trial. JAMA 288: 321-333.
90. Hodis HN, Mack MJ, Lobo RA, et al, 2001 Estrogen in the prevention of atherosclerosis; a randomized double-blind, placebo-controlled trial. Ann Intern Med 135: 939-953.
91. Hodis HN, Mack MJ, Azen SP, et al, 2003 [for the Women’s Estrogen-Progestin Lipid-Lowering Hormone Atherosclerosis Regression Trial Research Group]. Hormone therapy and the progression of coronary artery atherosclerosis in postmenopausal women. N Engl J Med 349: 535-545.
92. Wagner JD, Kaplan JR, Burkman RT, 2002 Reproductive hormones and cardiovascular disease; mechanism of action and clinical implications. Obstet Gynecol Clin N Am 29: 475-493.
93. Holm P, Andersen HL, Andersen MR, et al, 1999 The direct antiatherogenic effect of estrogen is present, absent or reversed, depending of the state of the arterial endothelium. Circulation 100: 1727-1733.
94. Mikkola TS, Clarkson TB, 2002 Estrogen replacement therapy, atherosclerosis and vascular function. Cardiovasc Res 53: 605-619.
95. Herrington DM, Espeland MA, Crouse JR, et al, 2001 Estrogen replacement and branchial artery flow-mediated vasodilation in older women. Arterioscler Thromb Vasc Biol 21: 1955-1961.
96. Bird AD, 1986 CpG-rich islands and the function of DNA methylation. Nature 321: 209-213.
97. Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al, 1999 Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res 43: 985-991.
98. Fergusan AT, Lapidus RG, Baylin SB, Davidson NE, 1995 Demethylation of the estrogen receptor gene in ER-negative breast cancer cells can re-activate estrogen receptor gene expression. Cancer Res 55: 2279-2283.
99. Clarkson TB, Anthony MS, Jerome CP, 1998 Lack of effect of raloxifene on coronary artery atherosclerosis of postmenopausal monkeys. J Clin Endocrinol Metab 83: 721-726.
100. Bjarnason NH, Haarbo J, Byrjalsen I, et al, 1997 Raloxifene inhibits aortic accumulation of cholesterol in ovariectomized, cholesterol-fed rabbits. Circulation 96: 1964-1969.
101. Barret Connor E, Grady D, Sashegyi A, Anderson PW, Cox DA, Hoszowski K, et al, 2002 for the MORE investigators. Raloxifene and cardiovascular events in osteoporotic postmenopausal women; 4-years’ results from the MORE randomized trial. JAMA 287: 847-857.
102. Genazzani AR, Benedek-Jaszmann LJ, Hart DM, et al, 1991 Org OD14 and the endometrium. Maturitas 13: 243-251.
103. Ginsburg J, Prevelic J, Butler D, et al, 1995 Clinical experience with tibolone [LIVIAL] over 8 years. Maturitas 21: 71-76.
104. Bjarnason NH, Bjarnason K, Haarbo J, et al, 1997 Tibolone; influence on markers of cardiovascular disease. J Clin Endocrinol Metab 82: 1752-1756.
105. Farish E, Barnes IF, Fletcher CD, et al, 1996 Effects of tibolone on serum lipoprotein and apolipoprotein (a) levels compared with a cyclical estrogen/progestogen regimen. Menopause 6: 98-104.
106. Klein OK, Janfaza M, Wong JA, Chang RJ, 2003 Estrogen bio-activity in fo-ti and other herbs used for their estrogen-like effects as determined by a recombinant cell bioassay. J Clin Endocrinol Metab 88: 4077-4079.
107. Anderson JJ, Anthony M, Cline JM, et al, 1999 Health potential of soy isoflavones for menopausal women. Public Health Nutr 2: 489-504.
108. Anderson JW, Johnstone BM, Cook-Newell ME, 1995 Meta-analysis of the effects of soy protein intake on serum lipids. N Engl J Med 333: 276-282.
109. Anthony MS, Clarkson TB, Bullock BC, et al, 1997 Soy protein versus soy phytoestrogen in the prevention of diet-induced coronary artery atherosclerosis of male cynomolgus monkeys. Arterioscler Thromb Vasc Biol 17: 2524-2531.
110. Beoglehale R, 1990 International trends in coronary heart disease mortality, morbidity and risk factors. Epidemiol Rev 12: 1-15.
111. Wagner JD, Cefalu WT, Anthony MS, et al, 1997 Dietary soy protein and estrogen replacement therapy improve cardiovascular risk factors and decrease aortic cholesteryl ester content in ovariectomized cynomolgus monkeys. Metabolism 46: 698-705.
112. Kronenberg F, Fugh-Berman A, 2002 Complementary and alternative medicine for menopausal symptoms; a review of randomized controlled trials. Ann Intern Med 137: 805-813.
113. Psaty BM, Smith ML, Lemuitre RN, et al, 2001 HRT; prothrombotic mutations and the risk of incident non-fatal myocardial infarction in postmenopausal women. JAMA 285: 906-913.