Abstract
Although paediatric Cushing's disease is rare, it is associated with severe morbidity in childhood and presents a major diagnostic and therapeutic challenge for the paediatric endocrinologist. Growth failure remains an important feature of paediatric Cushing's disease, both at diagnosis and after successful treatment. However, the development of specific diagnostic tests and important therapeutic advances has contributed significantly to the current management. Transsphenoidal pituitary surgery (TSS) is now accepted as first-line therapy for both adult and paediatric Cushing's disease, offering the best opportunity for cure while preserving normal pituitary function. The cure rate following TSS in our centre is currently 62%; therefore, a significant proportion of children following TSS will remain uncured. Our favoured second-line therapy is pituitary radiotherapy, which is effective, rapid and appears to spare anterior pituitary function, with the exception of GH secretion. Growth failure was a symptom at diagnosis in 74% of patients; mean height SDS -1.81 (-0.28 to -4.17) compared with mean BMI SDS 2.29 (1.72-5.06). Height velocity (HV) was also subnormal; range 0.9 – 3.8 cm/yr. Chronic hypercortisolaemia suppresses linear growth and a history of symptoms for >2 years was obtained in 72% of our paediatric patients with Cushing's disease. In our series, 9 out of 10 patients did not demonstrate catch-up growth 0.64 years after TSS or pituitary radiotherapy and peak serum GH level to glucagon/ITT stimulation was 0.5 – 20.9 mU/l. Assessment of GH secretion over a period of 6 to 108 months after cure of paediatric Cushing's disease suggests that impaired GH secretion may persist into adult life. Investigation of possible GHD is performed in our unit in all post-cure patients who do not show optimal catch-up growth. If GH secretion is subnormal, GH replacement is started early and continued until final height, when GH secretion is reassessed. In our series of 10 patients, 9 received GH therapy, combined in 3 with a GnRH analogue to arrest puberty. The results of this treatment were satisfactory with mean height deficit compared with target height improving significantly from mean -1.72 ± 1.26 SDS at diagnosis to -0.93 ± 1.13 SDS (p=0.005) at final height or latest assessment, indicating that a favourable final height can be achieved.
INTRODUCTION
Paediatric Cushing's disease is rare, but is nevertheless associated with severe morbidity and presents a major diagnostic and therapeutic challenge for the paediatric endocrinologist. There are few large series of paediatric patients with Cushing's disease in the literature. Of those that exist, the publications by the group of Chrousos at NIH remain pre-eminent1 and clearly describe the state-of-the-art approaches to current diagnostic and therapeutic strategies. As at NIH, we have embraced the concept of close paediatric and adult endocrinology co-operation in the management of this difficult disorder2. In fact, with the possible exception of the unit of Grumbach in San Fransisco3, single paediatric endocrine centres have had difficulty acquiring sufficient experience to be confident in managing paediatric Cushing's disease.
ADVANCES IN THE DIAGNOSIS OF CUSHING'S DISEASE
In the past 20 years, the development of three specific diagnostic techniques has contributed significantly to the current management of Cushing's disease. These are the CRH test, state-of-the-art radiological imaging of the pituitary gland and bilateral inferior petrosal sinus catheterisation. The CRH test, which demonstrates an exaggerated serum cortisol response in patients with Cushing's disease, is now accepted as part of the protocol for investigation of possible Cushing's syndrome in both adults and children1,2,4-6. Pituitary imaging is also routinely performed to attempt to identify a corticotroph microadenoma. In the paediatric NIH series, 52% of patients were reported to have an abnormal pituitary MRI consistent with the presence of an adenoma1. We have found pituitary imaging relatively unhelpful2-7 with <10% of paediatric patients demonstrating an abnormality, which was subsequently identified at surgery to be the microadenoma responsible for Cushing's disease. Nevertheless, high quality pituitary imaging remains a potentially important and necessary investigation.
Bilateral inferior petrosal sinus catheterisation with sampling for ACTH (IPSS) was developed at NIH1-8 and is now performed in most centres of adult endocrinology managing significant numbers of patients with Cushing's disease4. Its practice in children has been slower in becoming established, mainly because
of lack of experience due to the rarity of paediatric Cushing's disease. We have now performed IPSS as part of the diagnostic protocol in over 15 paediatric patients with Cushing's disease. Localisation of the microadenoma, by demonstration of an interpetrosal sinus ACTH gradient of >1.4 indicating lateralisation of hormone secretion, is of value and directly linked to successful outcome of TSS7.
CURRENT THERAPEUTIC APPROACH IN PAEDIATRIC CUSHING'S DISEASE
A number of important therapeutic advances have occurred in the past two decades. Perhaps the most significant is the development of transsphenoidal pituitary surgery (TSS). This surgical technique is now accepted as the first choice of therapy for both adult and paediatric Cushing's disease1,9-11. Although technically difficult, particularly in children, removal of the corticotroph microadenoma responsible for Cushing's disease offers the best opportunity for cure while retaining normal pituitary function.
Following TSS, even in the most skilled hands, a significant proportion of children will remain uncured, when a post-operative cortisol value of <50 nmol/l is considered a key criterion of cure6-12. Currently in our centre, in 21 children and adolescents with Cushing's disease, the cure rate is 62% following TSS5. Three possible second-line therapies exist after unsuccessful TSS. These are: repeat TSS, pituitary radiotherapy and bilateral adrenalectomy. Our favoured choice is pituitary radiotherapy, which is effective, rapid and appears to spare anterior pituitary function, with the exception of GH secretion13,14. Bilateral adrenalectomy is associated with risk of Nelson's syndrome2,15,16 and is therefore currently not recommended.
GROWTH AT DIAGNOSIS OF CUSHING'S DISEASE IN CHILDREN
It is well recognised that chronic hypercortisolaemia suppresses linear growth1,17,18. In 19 paediatric patients with Cushing's disease, growth failure was volunteered as a symptom at diagnosis in 74%, with a mean height SDS of -1.81 (-0.28 to -4.17)5. This contrasted strikingly with a mean BMI SDS of 2.29 (1.72 – 5.06). Height velocity was subnormal, ranging from 0.9 – 3.8 cm/year. The broad range of height SDS values (Figure 1) shows that height at diagnosis is not always abnormal and is probably related to the length of history. Interestingly, we have found that parents frequently do not realise that their child is developing Cushing's syndrome. This often leads to a long duration of symptoms before medical attention is sought and the diagnosis is made. A history of symptoms for >2 years was obtained in 72% of our patients5. Consequently, there is frequently a protracted period of symptomatology during which short stature can develop.
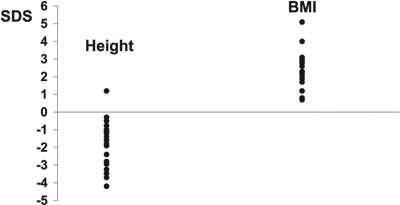
Figure 1. Height SDS and body mass index (BMI) SDS values at diagnosis in 19 patients with paediatric Cushing's disease.
The mechanism of growth failure in Cushing's disease is complex. It has been suggested that there is reduced bioactivity of IGF-I19, although serum IGF-I levels are usually normal18,19. Magiakou elegantly demonstrated reduced physiological GH secretion at diagnosis19. We do not routinely assess GH secretion before treatment.
Adrenal androgens may be normal or elevated in children with Cushing's disease. In a small series of patients, levels of androstenedione and DHEA-S, after conversion to SD scores, were not significantly abnormal20. However, virilization is frequent at diagnosis indicating increased exposure to androgens. Bone age was delayed in 14/16 patients at diagnosis20. This suggests that hypercortisolaemia has a predominant delaying effect on skeletal maturation, compared with adrenal androgens, and many children and adolescents have potential for catch-up growth after cure of their Cushing's disease.
EFFECT OF TREATMENT OF PAEDIATRIC CUSHING'S DISEASE ON GH SECRETION
Successful treatment by TSS can be associated with GH deficiency. This was demonstrated in the paediatric NIH series19 and in adults18. Adults, however, recovered normal GH secretion 2 years after cure of Cushing's disease18. We have reported that GH deficiency was present in the majority of patients who were successfully treated. This was the case during the first year after cure by TSS21 or by pituitary radiotherapy14 (Figure 2). Assessment of GH secretion over a longer period of 6 to 108months after cure of paediatric Cushing's disease has recently been performed22. Out of 13 patients, normal GH secretion, i.e. peak GH >30 mU/l, was only documented in 3, suggesting that impaired GH secretion may well persist into adult life, although it may not be severe enough to justify GH replacement therapy.
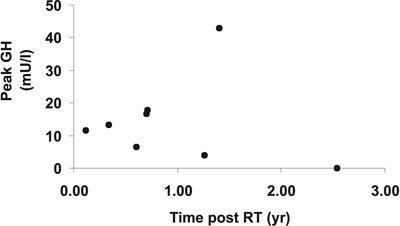
Figure 2. Peak GH levels after stimulation with ITT or glucagon in 9 patients with paediatric Cushing's disease treated with pituitary radiotherapy.
OPTIMIZATION OF LINEAR GROWTH AFTER CURE OF PAEDIATRIC CUSHING'S DISEASE
The normalization of growth and final adult height in patients cured of Cushing's disease in childhood presents a major challenge to the paediatric endocrinologist. One might expect spontaneous catch-up growth to occur after cure, when hypercortisolaemia is no longer present. However, post-operative growth may be disappointing with a sub-optimal pubertal growth spurt and failure to achieve normal adult height17,19. Our experience agrees with this finding since during an interval of 0.64 years after TSS or pituitary radiotherapy, 9 out of 10 patients in our series failed to show clear evidence of catch-up growth21. These patients were therefore tested for GH deficiency, which was present to some degree (peak GH to glucagon/ITT stimulation 0.5 – 20.9 mU/l) in all 9. Growth hormone deficiency was also demonstrated in the patients who were treated by pituitary radiotherapy14.
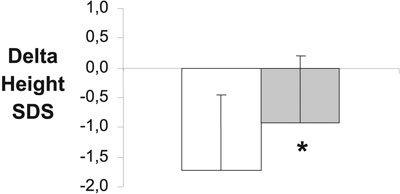
Figure 3. Delta height SDS (height SDS – target height SDS) in 10 patients with paediatric Cushing's disease at presentation (left panel) and at final height or latest assessment (right panel). * P=0.005.
CONCLUSIONS
Growth failure is an important feature of paediatric Cushing's disease, both at diagnosis and after successful treatment. From the diagnostic point of view, the contrast between subnormal height and excessive weight at the time of presentation would favour a diagnosis of Cushing's syndrome. It should be remembered that most children with simple obesity have tall rather than short stature. After successful cure, GH status should be urgently assessed. We would advise against a prolonged period of observation, based on the assumption that optimal spontaneous catch-up growth will occur. In our experience this is infrequent. The combination of sub-normal GH response to provocation and sub-optimal catch-up growth is, in our view, a basis for prompt GH replacement, combined in pubertal patients with GnRH analogue therapy. Long-term growth and final adult height on this regimen has not yet been formally assessed; however, the early results are encouraging21 and suggest that a final adult height within the parental target is likely to be achieved.
This paper is based on a lecture, delivered by Prof. M. Savage on the occasion of the 7th annual meeting of the Hellenic Society for the Study and Application of Growth Hormone, held on 7 December 2002.
REFERENCES
1. Magiakou MA, Mastorakos G, Oldfield EH, et al, 1994 Cushing's syndrome in children and adolescents, presentation, diagnosis and therapy. New Engl J Med 331: 629-636.
2. Weber A, Trainer PJ, Grossman AB, et al, 1995 Investigation, management and therapeutic outcome in 12 cases of childhood and adolescent Cushing's syndrome. Clin Endocrinol 43: 19-28.
3. Styne D, Grumbach M, Kaplan S, Wilson C, Conte F, 1984 Treatment of Cushing's disease in childhood and adolescence by transsphenoidal microadenomectomy. New Engl J Med 310: 889-893.
4. Newell-Price J, Morris DG, Drake WM, et al, 2002 Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab 87: 1640-1645.
5. Savage MO, Lienhardt A, Lebrethon MC, et al, 2001 Cushing's disease in childhood: presentation, investigation, treatment and long-term outcome. Horm Res 55: 24-30.
6. Newell-Price J, Trainer PJ, Besser GM, Grossman AB, 1999 The diagnosis and differential diagnosis of Cushing's and pseudo-Cushing's states. Endocr Rev 19: 647-672.
7. Lienhardt A, Grossman AB, Dacie JE, et al, 2001 Relative contributions of inferior petrosal sinus sampling and pituitary imaging in the investigation of children and adolescents with ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab 86: 5711-5714.
8. Oldfield EH, Doppman JL, Nieman LK, et al, 1991 Petrosal sinus sampling with corticotropin-releasing hormone to distinguish Cushing's disease from pseudo-Cushing states or normal physiology. N Engl J Med 325: 897-905.
9. Devoe DJ, Miller WL, Conte FA, et al, 1997 Long term outcome in children and adolescents after transsphenoidal surgery for Cushing's disease. J Clin Endocrinol Metab 82: 3196-3202.
10. Fahlbusch R, Honegger J, Buchfelder M 1994 Neurosurgical management of Cushing's disease in children In: Savage MO, Bourguinon J, Grossman AB (eds) Frontiers in paediatric endocrinology, Blackwell Scientific Publications, Oxford; pp, 68-72.
11. Ludecke D, 1990 Transnasal microsurgery of Cushing's disease 1990. Overview including personal experiences with 256 patients. Pathol Res Pract 187: 608-612.
12. Trainer PJ, Lawrie HS, Verhelst J, et al, 1993 Transsphenoidal resection in Cushing's disease: undetectable serum cortisol as the definition of successful treatment. Clin Endocrinol 38: 73-78.
13. Jennings A, Liddle G, Orth D, 1977 Results of treating childhood Cushing's disease with pituitary irradiation. N Engl
J Med 297: 957-962.
14. Storr H, Plowman PN, Carroll P, Savage MO, Krassas GE, 2003 Clinical and endocrine responses to pituitary radiotherapy in paediatric Cushing's disease: an effective second line treatment. J Clin Endocrinol Metab 88: 34-37.
15. Hopwood N, Kenny F, 1979 Incidence of Nelson's syndrome after adrenalectomy for Cushing's syndrome. Am J Dis Childhood 131: 1353-1356.
16. McArthur R, Hayles A, Salassa R, 1979 Childhood Cushing's disease: results of bilateral adrenalectomy. J Pediatr 95: 214-219.
17. Magiakou MA, Mastorakos G, Chrousos G, 1994 Final stature in patients with endogenous Cushing's syndrome. J Clin Endocrinol Metab 70: 1082-1085.
18. Hughes N, Lissett C, Shalet S, 1999 Growth hormone status following treatment of Cushing's disease. Clin Endocrinol 51: 61-66.
19. Magiakou MA, Mastorakos G, Gomez M, Rose S, Chrousos G, 1994 Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing's disease before and after surgical cure. J Clin Endocrinol Metab 78: 131-137.
20. Ho J, Blair J, Perry LA, et al, 2001 Evolution of serum androstenedione, DHEA-S, testosterone and SHBG concentrations at diagnosis and following treatment of paediatric Cushing's disease (CD). Red Res 49: 121.
21. Lebrethon M-C, Grossman AB, Afshar F, Plowman PN, Besser GM, Savage MO, 2000 Linear growth and final height after treatment for Cushing's disease. J Clin Endocrinol Metab 85: 3262-3265.
22. Carroll P, Monson J, Grossman AB, et al 2003 Successful treatment of childhood-onset Cushing's disease is associated with persistent reduction in growth hormone secretion. Clin Endocrinol (in press).
Address correspondence and requests for reprints to:
Gerasimos E. Krassas, Associate Professor of Medicine,
Department of Endocrinology and Metabolism, Panagia General Hospital,
22 N. Plastira str., N. Krini, 55132 Thessaloniki, Greece, Tel.: +30 2310 278279,
Fax: +30 2310 447444, e-mail: krassas@the.forthnet.gr
Received 24-12-02, Revised 31-01-03, Accepted 14-02-03