HORMONES 2004, 3(2):127-131
DOI: 10.14310/horm.2002.——
Address correspondence and requests for reprints to:
Prof. Ashley B. Grossman, Dept. of Endocrinology St. Bartholomew’s Hospital, London EC1A 7BE, United Kingdom, Tel: 207-6018343, Fax: 207-6018505, E-mail: A.B.Grossman@qmul.ac.uk
Received 26-01-04, Revised 02-03-04, Accepted 06-03-04
Abstract
Phaeochromocytomas (PC) and paragangliomas are disorders of the sympatho-adrenomedullary system. They are chromaffin-containing neuroendocrine tumors of neural crest origin that contain catecholamine-secreting granules: they arise from either the adrenal medulla (phaeochromocytomas) or from extra-adrenal neural crest derivatives e.g. the sympathetic chain (paragangliomas). The term paraganglioma is also used for vascular head and neck tumors derived from parasympathetic tissue, which commonly arise at the carotid bifurcation. It has been reported that some 10% of phaeochromocytomas are part of a familial syndrome, although recent data have suggested that germline mutations in known predisposing syndromes, such as multiple endocrine neoplasia type 2 (MEN2) and Von Hippel-Lindau (VHL), occur in a much higher percentage. However, familial genetic syndromes have been said to be less common in paragangliomas, although more recently described genetic syndromes may not have been considered. Thus, there is increasing evidence that mutations of subunits of the succinate dehydrogenase gene (SDHB, SDHC & SDHD) may confer susceptibility to paragangliomas and head-and-neck paragangliomas (HNPGL). We report a case of a patient with a previously published gene mutation in SDHB who had a single paraganglioma arising from the bladder with a characteristic clinical presentation, and in whom there was a positive family history of a HNPGL. He has demonstrated malignant recurrence with metastases which have been treated, so far successfully, with radiolabelled MIBG.
Key words: Paraganglioma, Phaeochromocytoma, Malignant, Bladder, Succinate dehydrogenase, Carotid body tumor, MIBG therapy
INTRODUCTION
The most common type of catecholamine-secreting tumors is a phaeochromocytoma. Around 10% of these tumors, however, arise from structures outside of the adrenal gland, most commonly the sympathetic chain on either side of the aorta, and usually below the diaphragm. Occasionally such tumors, referred to as extra-adrenal phaeochromocytomas or paragangliomas, can be extremely difficult to localise, and complex imaging techniques have evolved to attempt to locate them. It is being increasingly recognised that a substantial minority of both phaeochromocytomas and paragangliomas are familial, and the genetic basis for these tumors is gradually being determined. However, the long-recognised association of phaeochromocytomas with multiple endocrine neoplasia type 2 (MEN 2) and Von Hippel Lindau syndrome (VHL) tends to occur in older patients, and until recently the genetic basis for paragangliomas has been unclear. We present a detailed clinical description of a patient in whom careful history taking revealed the unexpected finding of a paraganglioma in an unusual location, and in whom the presence of a relevant family history led to detailed and ultimately successful attempts to identify a germline susceptibility mutation briefly mentioned in a recent publication1. In addition, this case illustrates an approach to therapy in a young patient in whom the tumor was found to be malignant.
CASE REPORT
The patient first presented to his general practitioner in 1997 aged 24 years with episodic severe `thumping’ headaches radiating to the eyes and the teeth and lasting up to approximately 25 minutes at a time; the patient had noted that they were associated with sweating and palpitations. He was then referred to hospital where he was investigated by an endocrine team. At that time the history was as noted previously, but it was now clear that the majority of the headaches occurred first thing each morning, shortly after the patient had risen from bed. On each occasion he had noted that the pain in his head occurred some 3-5 minutes after he had voided his bladder. There were no additional clinical symptoms and the rest of the systematic enquiry was negative. The patient had no significant past medical history. However, in his family history it was recorded that his mother had died at age 32y of what he believed was referred to as a carotid body tumor. Examination revealed a fit, slim young man with a regular pulse of 50 beats per minute and blood pressure 130/85 mmHg supine.
It was suspected that he may have a catecholamine-secreting tumor in the region of his bladder, and baseline investigation confirmed significantly elevated urinary noradrenaline of 1230, 1340 and 1090 nmol/24hr (upper 99% confidence limit = 814 nmol/24hr); urinary adrenaline and dopamine excretion were normal. The patient had an MRI chest/abdomen and ultrasound (USS) of his thyroid and carotid bodies; the positive finding was a tumor in the anterior superior wall of the bladder revealed by the MRI (Figure 1, A & C).
An USS of the bladder was performed which revealed a 4cm lesion in the bladder (Figure 1, B). A 123I-mIBG radionuclide scan revealed increased uptake in the right side of the bladder.
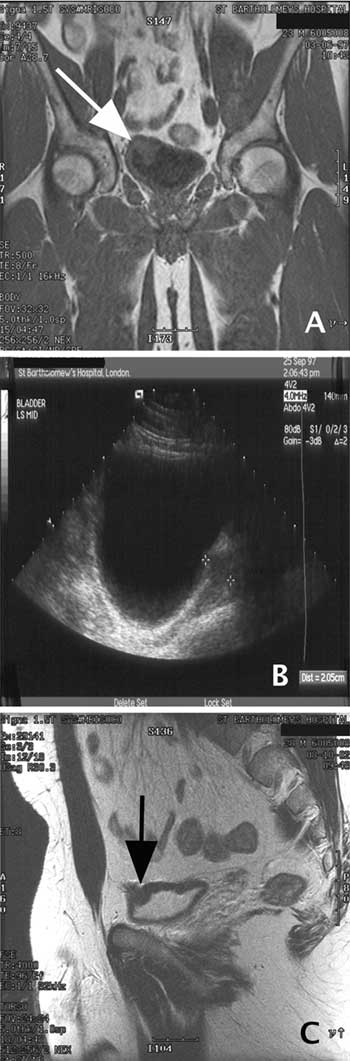
Figure 1. A. A T2-weighted coronal MRI scan of the abdomen showing lesion in right upper wall of bladder (white arrow). B. USS of bladder, showing a lesion between the cross-hairs. C. A T1-weighted sagittal MRI scan of the abdomen showing the lesion in right upper wall of bladder (black arrow).
The tumor was surgically excised by cystoscopy soon after diagnosis and the surgery was considered curative. The histology revealed a typical paraganglioma with some evidence of invasiveness, but no tumor at the excision margin. The patient was discharged asymptomatic. Three months later he was routinely reviewed and he remained well. He then continued to remain well between 1997 and 2001. However, in 2001 he re-presented with `severe’ headaches which were `different from four years previously’. They were described as sharp, occipital and left-sided, and the teeth, throat and gums were involved, but there was no association with micturition this time.
Investigations at this time showed normal urinary catecholamines but 123I-mIBG uptake in the region of the right shoulder, sternum and mediastinum, and the right upper pole of the bladder. An MRI scan showed lymphadenopathy related to the previous bladder lesion, but there were no plain radiology or CT correlates of the other MIBG-avid lesions. He was therefore diagnosed with possible recurrence of his bladder paraganglioma with metastatic disease, and was started prophylactically on á-adrenoceptor blockade with doxazosin 4mg od. In October 2002 he was readmitted, with similar 123I-mIBG scanning results; however, an isotope bone scan showed uptake in the sternum and medial end of the clavicle, at the same sites of positive MIBG avidity and compatible with metastatic disease, while MRI of the pelvis demonstrated progression of the bladder disease. Urinary catecholamines were now elevated once more (urinary noradrenaline 938 and 1091 nmol/24hr). He was therefore treated with a therapy dose of 200 mCi (c.7 GBq) of 131I-mIBG, and this was repeated without adverse effects 6 and 12 months later. Post-therapy scans showed uptake similar to the diagnostic scan. He will continue to be treated up to a total of 6 doses of mIBG, barring any adverse event.
In view of the positive family history, genetic screening was undertaken, but no mutations of the relevant coding regions of the MEN2 (ret-proto-oncogene) and VHL loci were demonstrated and there was no clinical evidence of neurofibromatosis type 1. However, in view of the recent association of germline mutations of the mitochondrial complex enzyme succinate dehydrogenase (SDH) with head-and neck paragangliomas (HNPGL), we investigated genomic DNA for the B, C and D subunits of this enzyme. We identified an SDH subunit B mutation in this patient. This was a non-conservative nucleotide change in the coding region c.590C>G, associated with an amino-acid change P197R. This patient had presented with a paraganglioma at the age of 24, while his mother had died with an HNPGL at age 32 years. Germline mutations in SDHB are more commonly associated with phaeochromocytoma and paraganglioma than HNPGL, whereas SDHD mutations are more commonly associated with HNPGL.
DISCUSSION
Bladder paragangliomas have a very characteristic clinical presentation and a good history can lead to a rapid diagnosis of an important condition. The most defining complaint in this patient was that the headaches appeared to increase in intensity upon micturition. The presence of a catecholamine-secreting tumor was confirmed by the presence of an elevated urinary noradrenaline, although this was not grossly increased and this level of elevation can be found in a number of other conditions. However, in this case both MR and USS confirmed the presence of a bladder mass which, together with the clinical history and urinary noradrenaline level, rendered the diagnosis of a bladder paraganglioma highly probable. Uptake by radiolabelled mIBG, which is of only moderate sensitivity but of high specificity for the diagnosis of a catecholamine-secreting tumor, confirmed the diagnosis.
Catecholamine-secreting tumors may arise either de novo or as a component of certain inherited cancer syndromes. The most common syndromes include VHL and MEN type 2. These syndromes are caused by a germline mutations in the VHL tumor suppressor gene and the RET proto-oncogene, respectively. Neurofibromatosis type 1 is a third syndrome which determines susceptibility to phaeochromocytoma, although it is only seen in 1% of NF-1 patients. According to established thinking, around 10% of phaeochromocytomas have been considered to be familial, but this has recently been challenged and reports of familial prevalence of apparently sporadic tumors have ranged up to 24%2. Previous genetic investigations have focussed on VHL and RET, but recent findings that germline mutations in SDH subunits (particularly B and D) predispose to families with inherited HNPGL has raised increasing interest in these SDH subunit mutations as important factors in apparently sporadic tumors.
Paragangliomas can be sympathetic in origin (mainly localised to the retro-peritoneum and thought to arise from residual rests of chromaffin tissue around the aorta, especially the bifurcation of the aorta _ the “organs of Zuckerkandl”) or parasympathetic, with the aortic arch and the neck being important sites for these tumors. The most common site for a HNPGL is the carotid body, but these tumors are usually non-functioning tumors, unlike the secreting tumors of the adrenal medulla. Succinate Dehydrogenase (SDH), which encodes mitochondrial complex II, is composed of four subunits, SDHA, SDHB, SDHC & SDHD. SDHA & SDHB are the enzymatic subunits, while SDHC & SDHD anchor the complex to the mitochondrial membrane. Hereditary head and neck paragangliomas that target the carotid body (HNPGL) are associated with a mutation in the gene encoding SDHD3,4.
An important feature in this patient’s history was the familial association with HNPGL. SDHD loss-of-function mutations have been shown to predispose to HNPGL and phaeochromocytomas and paragangliomas, so patients with phaeochromocytoma or paraganglioma and SDHD mutations should be regarded as at risk for carotid body tumors. Familial paraganglioma is inherited as a dominant trait with parent-of-origin effects such that paternally transmitted mutations cause disease whereas maternal transmission is not associated with clinical disease2,3. The finding that germline mutations of SDHD cause hereditary and apparently sporadic paragangliomas implies a tumor-suppressor function for SDH, and it may be that there is faulty oxygen sensing and responsiveness, as is thought to be aetiological in the substrate for the VHL gene. For reasons which are unclear, SDHD mutations are more commonly associated with the development of HNPGL, whereas SDHB have been more
frequently coupled with phaeochromocytoma susceptibility1,5,6 (the patient is case K16 in reference 1).
The role of SDH in coding for mitochondrial complexes, and hence a key involvement in the TCA cycle and aerobic electron transport chain of mitochondria, raises questions about the link between mitochondria (and mutations of its complex subunits) and hypoxia. A marked increase in the incidence of carotid body tumors is seen in people at high altitude, where there is `relative’ hypoxia compared to those living at sea-level. The carotid body is linked to oxygen sensing and serves a chemosensory role. Chronic hypoxia exposure at high altitudes has been shown to be associated with neoplastic enlargement of the carotid body. As chronic hypoxia has been shown to predispose to neoplastic enlargement of the carotid body, and as mitochondrial complex II is intimately involved hypoxia and oxidation-reduction, it is believed that defects in MC II could be pathogenetic in carotid glomus tumors and familial paraganglioma and phaeochromocytoma.
Finally, our case illustrates another modality of therapy available when there is metastatic disease. In those cases where the tumor recurs or is metastatic at presentation, or there is a tumor which has advanced beyond surgery, targeted radionuclide therapy can be used to retard the progression of the tumor and increase the quality of life. These tumors are relatively chemoresistant, and 131I-mIBG is relatively free of adverse effects and easy to administer7-9. Whether it will lead to permanent inhibition of tumor growth remains unclear, but in general there are few permanent cures following this type of therapy, although long-term remission is probable8. There is also evidence that tumors in patients with SDHB mutations run a more indolent course than might be expected when malignant10.
In summary, we present a case of a bladder paraganglioma with a typical clinical presentation in whom the family history led to the search for a germline mutation, which was established to occur in the B subunit of the mitochondrial enzyme SDH. The presence of metastatic disease has led to regular therapy with 131I-mIBG, so far successfully.
REFERENCES
1. Astuti D, Hart-Holden N, Latif F, et al, 2003 Genetic Analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and phaeochromocytoma susceptibility. Clin Endocrinol 59: 728-733.
2. Maher ER, Eng C, 2002 The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Gen 20: 2347-2354.
3. Astuti D, Douglas F, Lennard TWJ, et al, 2001 Germline SDHD mutation in familial phaeochromocytoma. Lancet 357: 1181-1182.
4. Cascon A, Ruiz-Llorente S, Cebrian A, et al, 2002 Identification of novel SDHD mutations in patients with phaeochromocytoma and/or paraganglioma. Eur J Hum Gen 10: 457-461.
5. Astuti D, Latif F, Dallol A, et al, 2001 Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial phaeochromocytoma and to familial paraganglioma. Am J Hum Gen 69: 49-54.
6. Benn DE, Croxson MS, Tucker K, et al, 2003 Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene 22: 1358-1364.
7. Mukherjee JJ, Kaltsas GA, Islam N, et al, 2001 Treatment of metastatic carcinoids, phaeochromocytomas, paragangliomas and medullary carcinoma of the thyroid with 131I-metaiodobenzylguanidine. Clin Endocrinol 55: 47-60.
8. Kaltsas GA, Mukherjee JJ, Foley R, Britton KE, Grossman AB, 2003 Treatment of metastatic phaeochromocytoma and paraganglioma with 131I-metaiodobenzylguanidine (MIBG). The Endocrinologist 13: 1-14.
9. Bomanji JB, Wong W, Gaze MN, et al, 2003 Treatment of neuroendocrine tumors in adults with 131I-MIBG therapy. Clin Oncol (R Coll Radiol) 15: 193-198.
10. Young AL, Baysal BE, Deb A, Young WF, 2002 Clinical Case Seminar: Familial malignant catecholamine-secreting paraganglioma with prolonged survianl associated with mutation in the succinate dehydrogenase B gene. J Clin Endocrinol Metab 87: 4101-4105.