Abstract
A diagnosis of central hypothyroidism (CH) can be missed easily or delayed without a high index of suspicion due to normal or slightly altered thyroid stimulating hormone (TSH) levels during the initial screening test for thyroid dysfunction. A correct diagnosis of CH is very important for safely treating patients. Specifically, doctors must ensure a proper evaluation of combined adrenal insufficiency to prevent a fatal adrenal crisis. Here we report a case of CH combined with secondary adrenal insufficiency in a 42-year-old woman with Dyke-Davidoff-Masson syndrome, which is a rare neurological disease
INTRODUCTION
Central hypothyroidism (CH) is a disorder caused by defects in thyroid stimulating hormone (TSH) or TSH-releasing hormone (TRH) synthesis, despite a normally functioning thyroid gland.1 CH can be familial when caused by genetic mutations such as TSHβ, PAX8, TRH receptor, Pit-1 and Prop-1 or can arise sporadically with injuries to the hypothalamus or pituitary gland.2
Dyke-Davidoff-Masson syndrome (DDMS) is a rare disease characterized by cerebral hemiatrophy following intrauterine or perinatal damage to the developing brain3-5. There are few reports describing hypothalamic or pituitary function in patients with DDMS. Here we report a case of centrally originating hypothyroidism and adrenal insufficiency in a patient with DDMS.
CASE
A 42-year-old woman presented with nausea and dizziness, which were precipitated by an increased dose of antiepileptic drugs (phenytoin and phenobarbital). She had experienced meningitis at 12 months of age, which was followed by a left hemiparesis. We were unable to obtain the patient’s childhood medical records; thus, our clinical history relied on the records provided by the neurologist, who had recently begun to treat her, and the patient’s mother. The patient was diagnosed with a seizure disorder and had been given anti-seizure medication since the age of 7 years. On admission, she was 148 cm tall and weighed 73.8 kg. Her initial vital signs were a blood pressure of 100/70 mmHg, a pulse of 70 beats/min, and a respiratory rate of 20 breaths/min.
The laboratory findings included normal serum electrolytes and liver and renal function tests. Her phenytoin level was 19.9 μg/ml (therapeutic range 5-20) and her phenobarbital level was 30.6 μg/ml (therapeutic range 10–30). We found right calvarial thickening, right hyperpneumatization in the mastoid air cells, ipsilateral hemicerebral atrophy and cerebral peduncles with prominent cortical sulci, and hypoplasia of the basal ganglia and thalamus (Figure 1), which were suggestive of DDMS. Computed tomography (CT) angiography revealed hypoplasia of the right internal carotid artery and the right middle cerebral artery (Figure 1H). However, sagittal magnetic resonance imaging (MRI) revealed no definite abnormalities in the hypothalamus and showed a normal pituitary gland (Figure 1I). Thyroid function tests included a low serum triiodothyronine (T3) level (77.86 ng/dl, normal range 87-178) and low free thyroxine (T4) (0.49 ng/dL, normal range 0.58-1.64); however, TSH was 1.31 mU/L (normal range 0.34-5.60) in the absence of thyroid peroxidase (TPO) and thyroxine-binding globulin antibodies. Ultrasonography revealed that the thyroid gland was of normal size and structure, with homogeneous echogenicity, except for a 7-mm simple cyst. The patient was referred to our Endocrine Division because of the abnormal thyroid function. The patient’s clinical findings, including the history of a postnatal neurologic injury, no compensatory rise in TSH levels, and normal thyroid structure without thyroid autoantibodies, raised a suspicion of CH rather than primary hypothyroidism. Therefore, a combined pituitary function stimulation test with an intravenous injection of regular insulin (0.15 U/kg), TRH (400 μg), and luteinizing-hormone-releasing hormone (LHRH; 100 μg) was performed (Table 1). This showed a normal rise in TSH, with decreased cortisol and growth hormone (GH) responses. To determine the origin of the hormonal deficiency, 1 g/kg body weight of corticotropin-releasing hormone (CRH) was administered; the adrenocorticotropic hormone (ACTH) response was normal, suggesting normal pituitary function and a possible hypothalamic defect (Table 2).
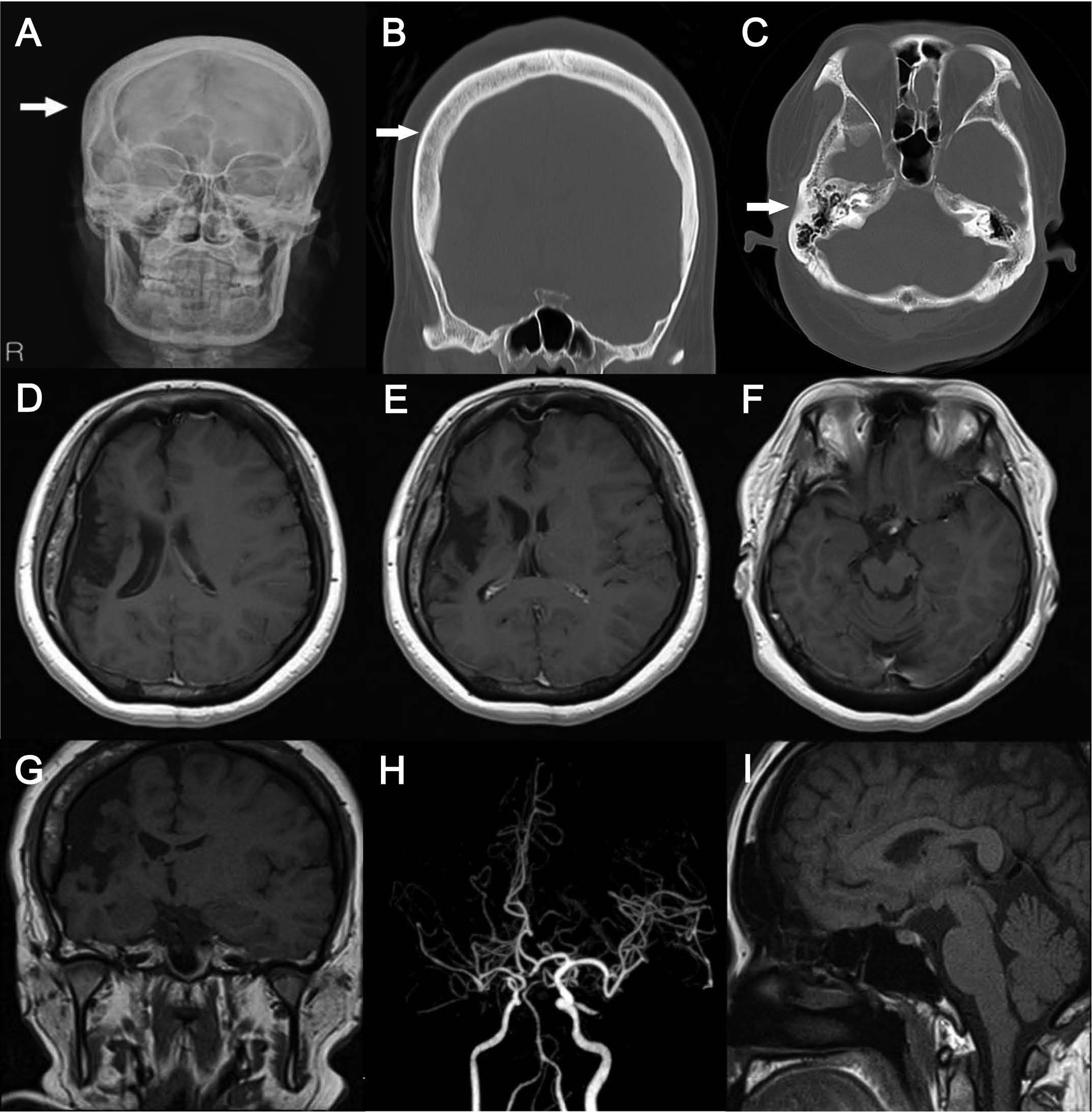
Figure 1. Skull x-ray (A) and computed tomography (CT) coronal sections (B) revealed right calvarial thickening, and CT axial sections (C) showed hyperpneumatization in the mastoid air cells. Brain magnetic resonance imaging (MRI) of axial (D-F) and coronal (G) sections showed diffuse atrophy involving the parietal, temporal, and part of the occipital lobes of the right hemisphere, cerebral peduncles with prominent cortical sulci on the ipsilateral side, and hypoplasia of the basal ganglia and thalamus, which
were suggestive of the Dyke- Davidoff-Masson syndrome. CT angiography (H) revealed hypoplasia of the right internal carotid artery and right middle cerebral artery. A sagittal MRI scan (I) revealed no definite abnormalities in the hypothalamus and showed a normal pituitary gland.
DISCUSSION
Dyke et al6first described DDMS in 1933 after observing the radiological features of nine patients with cerebral hemiatrophy combined with ipsilateral ventricular dilatation, compensatory osseous hypertrophy, and hyperpneumatization of the ipsilateral mastoid cells and paranasal sinuses.7 The clinical features of DDMS vary and include seizures, contralateral hemiparesis, facial asymmetry, and mental retardation according to the extent of brain injury.6 DDMS may be congenital or acquired from various etiologies.3-5,9 The intrauterine cause of the congenital form is generally unknown; however, cases with vascular abnormalities10 and coarctation of the midaortic arch11 have been reported, suggesting involvement of vascular factors in the brain damage. The causes of acquired DDMS are variable and include infection, trauma, ischemia, and hemorrhage.12 We believed our patient had acquired DDMS because she developed neurological abnormalities after severe meningitis at 12 months of age. Furthermore, the radiological findings supported an acquired cause of the disease. The presence of prominent sulci suggested that the brain insult occurred after development of the cerebral parenchyma rather than during embryogenesis.4 Compensatory skull thickening and enlarged mastoid cells indicated that the brain insult had occurred before the patient was 3 years of age and prior to maturation of the calvarium.3 The patient developed late seizures at 7 years of age. We are unable to explain the time lag between meningitis at 12 months of age and the seizures at age 7; however, it may be related to poor development of the cerebral hemisphere causing a decrease in neural activity and synaptic plasticity as a result of low neurotrophin levels.13
Hypothyroidism refers to thyroid hormone deficiency that usually stems from primary thyroid failure caused by chronic autoimmune thyroiditis and, very rarely, from central dysfunction of the pituitary or hypothalamus.14 Although a TSH measurement is the preferred screening test for thyroid insufficiency, the diagnosis can be missed when relying on the TSH levels in patients with CH.15 CH can manifest with a wide range of TSH levels, including normal, low, and even higher ranges than the reference values and a slightly low free T4 with a mild degree of dysfunction. Specifically, hypothalamic CH shows impaired TSH bioactivity resulting in qualitative defects rather than quantitative insufficiency.16 Consequently, a high level of clinical suspicion is quite important for ensuring the correct diagnosis and proper treatment of patients with CH who might have multiple other pituitary or hypothalamic hormone deficiencies. To avoid precipitating an adrenal crisis, the hypothalamic-pituitary adrenal axis must be assessed in patients with CH before replacing thyroid hormone.
Few studies have investigated pituitary or hypothalamic function in DDMS. Park et al17 reported a case of DDMS with hypopituitarism involving growth and sex hormones. In our case, CH was suspected based on the normal TSH level despite low T3 and free T4 levels without evidence of primary thyroid dysfunction, such as an immature thyroid gland, destruction of the thyroid parenchyma, or serum autoimmune thyroid antibodies. The pituitary stimulation test revealed a combined defect in the secretion of growth hormone and cortisol, in addition to the thyroid hormone insufficiency of central origin. Although the LH response was slightly lower than normal, gonadal hormone deficiency could be ruled out because she menstruated regularly. We confirmed that the patient had hypothalamic hypothyroidism with an intact TSH response (suggesting normal pituitary function) based on the insulin-induced stimulation test. This was further supported by the intact pituitary function based on the ACTH response in the CRH stimulation test and by a suspicious lesion in the hypothalamus with hypoplasia of the surrounding structures and a morphologically normal pituitary gland.
Antiepileptic drugs can alter thyroid hormone homeostasis. Many studies have reported that phenytoin treatment can cause decreases in total T4, free T4, T3, and free T3 without causing an increase in TSH in most cases.18 Although the underlying mechanism is unclear, it is thought to be related to interference with thyroid hormone binding19 and accelerated thyroxine clearance caused by induced hepatic mono-oxygenase.20 Therefore, overt thyroid insufficiency might have been accelerated by the elevated phenytoin dose in our patient with mildly comprised hypothalamic-pituitary-thyroid axis.
In conclusion, clinicians should bear in mind that cases of DDMS can be associated with hypothalamic or pituitary hormone deficiencies. A high index of clinical suspicion could increase the early diagnosis of CH and subsequently guide the proper assessment and treatment.
DISCLOSURE STATEMENT
The authors have nothing to declare as a conflict of interest.
RESULTS
1. Alexopoulou O, Beguin C, De Nayer P, Maiter D, 2004 Clinical and hormonal characteristics of central hypothyroidism at diagnosis and during follow-up in adult patients. Eur J Endocrinol 150: 1-8.
2. Grasberger H, Vaxillaire M, Pannain S, et al, 2005 Identification of a locus for nongoitrous congenital hypothyroidism on chromosome 15q25.3-26.1. Hum Genet 118: 348-355.
3. Atalar MH, Icagasioglu D, Tas F, 2007 Cerebral hemiatrophy (Dyke-Davidoff-Masson syndrome) in childhood: clinicoradiological analysis of 19 cases. Pediatr Int 49: 70-75.
4. Aguiar PH, Liu CW, Leitão H, et al, 1998 MR and CT imaging in the Dyke-Davidoff-Masson syndrome. Report of three cases and contribution to pathogenesis and differential diagnosis. Arq Neuropsiquiatr 56: 803-807.
5. Zilkha A, 1980 CT of cerebral hemiatrophy. AJR Am J Roentgenol 135:259-262.
6. Dyke CG, Davidoff LM, Masson CB, 1933 Cerebral hemiatrophy and homolateral hypertrophy of the skull and sinuses. Surg Gynecol Obstet: 588-600.
7. Ünal O, Tombul T, Cirak B, Anlar O, Incesu L, Kayan M, 2004 Left hemisphere and male sex dominance of cerebral hemiatrophy (Dyke-Davidoff-Masson Syndrome). Clin Imaging 28: 163-165.
8. Hsin YL, Chuang MF, Shen TW, Harnod T, 2011 Temporo-spatial analyses define epileptogenic and functional zones in a case of Dyke-Davidoff-Masson syndrome. Seizure 20: 713-716.
9. Sener RN, Jinkins JR, 1992 MR of craniocerebral hemiatrophy. Clin Imaging 16: 93-97.
10. Parker CE, Harris N, Mavalwala J, 1972 Dyke-Davidoff-Masson syndrome. Five case studies and deductions from dermatoglyphics. Clinical Pediatrics 11: 288-292.
11. Stred SE, Byrum CJ, Bove EL, Oliphant M, 1986 Coarctation of the midaortic arch presenting with monoparesis. Ann Thorac Surg 42: 210-212.
12. Tasdemir HA, Incesu L, Yazicioglu AK, Belet U, Güngör L, 2002 Dyke-Davidoff-Masson syndrome. Clin Imaging 26: 13-17.
13. Ono K, Komai K, Ikeda T, 2003 Dyke-Davidoff-Masson syndrome manifested by seizure in late childhood: a case report. J Clin Neurosci 10: 367-371.
14. Devdhar M, Ousman YH, Burman KD, 2007 Hypothyroidism. Endocrinol Metab Clin North Am 36: 595-615.
15. Faglia G, Bitensky L, Pinchera A, et al, 1979 Thyrotropin secretion in patients with central hypothyroidism: evidence for reduced biological activity of immunoreactive thyrotropin. J Clin Endocrinol Metab 48: 989-998.
16. Persani L, Ferretti E, Borgato S, Faglia G, Beck Peccoz P, 2000 Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 85: 3631-3635.
17. Park SY LM, Kim JH, Kim SY, Shin JY, Shin YG, Chung CH, 2010 A case of dyke-davidoff-masson syndrome associated with hypopituitarism and diabetes mellitus. Korean J Med 79: 316-320.
18. Benedetti MS, Whomsley R, Baltes E, Tonner F, 2005 Alteration of thyroid hormone homeostasis by antiepileptic drugs in humans: involvement of glucuronosyltransferase induction. Eur J Clin Pharmacol 61: 863-872.
19. Oppenheimer JH, Fisher LV, Nelson KM, Jailer JW, 1961 Depression of the serum protein-bound iodine level by diphenylhydantion. J Clin Endocrinol Metab 21: 252 -262.
20. Larsen PR, Atkinson AJ, Wellman HN, Goldsmith RE, 1970 The effect of diphenylhydantoin on thyroxine metabolism in man. J Clin Inves 49: 1266-1279.
Address for correspondence:
Eun Sook Kim, MD, PhD, Division of Endocrinology, Department of Internal Medicine,
Incheon St. Mary’s Hospital, The Catholic University of Korea,
665 Bupyeong 6-dong, Bupyeong-gu, 403-720, Incheon, Korea.
E-mail: 13900@catholic.ac.kr
Received 08-07-2012, Accepted 02-11-2012