DOI: 10.14310/horm.2002.—-
Address correspondence and requests for reprints to:
Dr. K. Karavanaki, 2nd Pediatric Department, “Aghia Sophia” Children’s Hospital, Goudi, 11527, Athens, Greece
Received 13-12-01, Revised 10-02-02, Accepted 01-03-02
Abstract
A 6-year old girl was examined having two years previously presented a transient Cushing’s syndrome, followed by recurrent hyponatremia, attributed to inappropriate ADH secretion (SIADH). The brain MRI showed no abnormalities on repeated examinations, except for a suggestion of empty sella syndrome. During the past two years she also presented recurrent episodes of a prolonged febrile illness of unknown origin. All investigations related to infectious, autoimmune neoplastic diseases, histiocytosis-X or neurosarcoidosis were negative and the fever was characterized as central. The patient also presented episodes of tonic-clonic seizures, myoclonias and behavioral problems (alternating states of irritability, sleepiness and apathy, optic and hearing illusions and phobias) with or without hyponatremia. Her cerebrospinal fluid (CSF) examination was not indicative of encephalitis and the encephalogram (EEG) showed only slowing of background activity. At the age of 4.75 years she, started to have recurrent episodes of hypopnoea/apnoea with severe desaturation and hypercapnia, occasionally requiring intubation and ventilation. She also developed unilateral miosis corneal ulceration and bilateral ptosis (oculo-sympathetic paresis). Repeat brain MRI and CT scans of the mediastinum excluded organic causes of apnoeas and of oculosympathetic paresis, such as neuroblastoma or lymphoma. Furthermore, on a 24hour electrocardiogram recording, using power spectral analysis, significantly reduced heart rate variability was observed, by comparison with age-specific normal ranges. Thus the apnoeas, ptosis, miosis and temperature instability were attributed to autonomic dysfunction. During the same period, the patient presented significant growth retardation and growth hormone (GH) deficiency was confirmed during two provocative tests (peak GH levels: 3.1 and 2.9 ng/ml (normal>10). Moreover, thyrotropin (TSH) deficiency and persistent hyperprolactinemia were detected. She was started on hGH and thyroxine. She was also put on fluid restriction and increased sodium intake for her SIADH. She was advised to use O2 administration by mask in case of apnoeas. The child died at age 6 6/12 years as a result of apnoea during sleep. In conclusion: Multiple pituitary hormonal abnormalities, together with symptoms of autonomic neuropathy (apnoeas, ptosis, miosis, tachycardia, temperature instability) and encephalopathy (seizures, myoclonias and behavioral problems) developed in a 4-year old girl. The suggested diagnoses were: 1.Neurometabolic disorder, 2. Mitochondrial disorder, 3. Post infectious autoimmune process.
CASE HISTORY:
A 6-year old girl was admitted to the hospital with the following history. She was born after an uncomplicated pregnancy and normal delivery with no perinatal problems. Until the age of 4 years, she was described as a completely healthy and normally growing girl, aside from the usual infections for age. Her psychomotor development was also normal. In her family history, three maternal uncles were reported as having psychosis.
At the age of 4 years, she went to spend the summer with her grandmother in a rural area. During her stay there and without any apparent cause she developed polyphagia and became obese, gaining 11 kgs in a period of approximately 3 months (BW: 28 kgs >>97the centile) (Figure 1) and assumed a Cushinoid appearance.

Figure 1.Age 3 10/12 years. Body weight:>> 97th centile.Height at 50th centile. Cushinoid appearance.
Shortly thereafter, she started to present tonic-clonic seizures, associated with severe hyponatremia, while at times she presented polydipsia and polyuria. The laboratory findings during hyponatremia were indicative of inappropriate secretion of ADH1: Serum Sodium 114 mmol/L, Chloride 65 mmol/L, Osmolality 238 mmol/kg, Uric acid 29.7 £mol/L and ADH 11.9 pmol/L (normal <7.6). The urine sodium was 151 mmol/lt, chloride 145 mmol/lt and urine osmolality 223 mmol/kg.
The plasma ACTH was 11.9 pmol/L (normal 1.9-11.4) and the serum cortisol was 89.1 nmol/L (normal 16.0-68.9). Eight months later, her body weight was reduced by 12 kgs and remained stable for the subsequent two years (Figure 2). The plasma ACTH was 6.4 pmol/L and the serum cortisol was 30.3 nmol/L. Hyperprolactinemia was a persistent finding (serum prolactin values ranged from 36 to 40 µg/L).
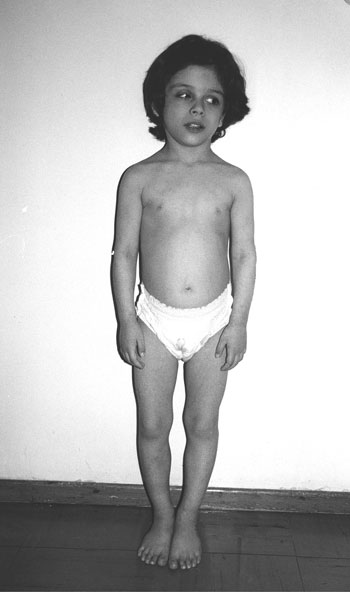
Figure 2. Age 6 2/12 years. Height: <3rd centile.Body weight: 10th centile.
A brain MRI did not disclose any abnormality, except for a suggestion of empty sella syndrome. The pituitary stalk and the characteristic bright spot of the posterior pituitary lobe were normal. Two subsequent brain MRI scans in the following two years did not disclose any abnormality.
A few months after the initial presentation, episodes of prolonged fever were added to the clinical picture, while her general condition was unaffected.
Blood, urine, cerebrospinal fluid (CSF) and stool cultures were negative. The ESR was 28 mm/1h, the CRP was 4 mg/dl (normal <3), the WBC was 7800/mm3 (Neutrophils: 57%, Lymphocytes: 21%) and the platelets 119.000/mm3. The CSF was negative for bacterial or viral infection and the CSF protein was 0.84 gr/l (normal: 0-0.3), the CSF lactate was 1.2 mmol/l t (normal: <2.5), the CSF glucose was 4.5 mmol/l and the CSF aminoacids were normal.
Other bacterial (Borrelia, toxoplasma Gondii, cryptococcus neophormans) and viral causes of infection (Epstein Barr virus and Enterovirus) were excluded by PCR or estimation of antibody levels in plasma or CSF. Immunoglobulins and sub-classes were normal and different autoantibodies were negative2. The neoplastic origin of prolonged febrile illness (neuroblastoma, lymphoma) was exluded by negative MRI and CT scan of the mediastinum, negative ultrasound of the abdomen and reduced levels of urinary concentration of vanillylmandelic acid (VMA), as well as the absence of hypertension.
Histiocytosis-X3 was also excluded by brain MRI, normal skull and long bones X-rays, normal chest CT scan and the absence of histiocytes in the CSF. Neurosarcoidosis4 was excluced by the normal levels of SACE (serum angiotensin converting enzyme:28-34 IU/lt (normal: 8-76)), normal brain MRI and chest CT scan5. Hence the fever was characterized as fever of unknown origin, or possibly central (hypothalamic).
The child subsequently presented phobias, introversion, aggressiveness, apathy, reduced reaction to pain and reduced appetite. A few months later the child was admitted to the Paediatric Intensive Care Unit (PICU) in a comatose state. During the subsequent 10 days, she manifested alternating states of irritability or sleepiness, while at times she was delusive (optic and hearing illusions). Despite impaired level of consciousness, her serum sodium level was normal. Her CSF examination was not indicative of encephalitis. She had an EEG (during sleep) which showed moderate excess of slowing of the background activity and was not indicative of encephalitis or epilepsy.
The child subsequently presented episodes of apnoea and loss of consciousness (coma stage I, Glasgow scale), at times requiring intubation and ventilation. She did not present bradycardia during severe hypoxia (SaO2: 60%, heart rate: 135 bpm). These episodes of apnoea gradually became quite frequent and were similar to cyanotic breath-holding attacks6. The child, although conscious, would not breathe, despite her severe hypoxemia and hypercapnia (hypoxia unawareness). Sleep studies were performed in order to further investigate the patient’s apnoeas. SaO2 was variable, with 9 dips per hour. Breathing was extremely irregular in rate and amplitude throughout the study and brief episodes of more rapid breathing suggested a principally neurological cause of her problems.
She was referred to Great Ormond Street Hospital, London, UK, for further investigation, but no conclusive diagnostic results were obtained.
During one episode of seizures, the child first presented miosis of the right pupil, ptosis bilaterally and corneal ulceration on the right eye, which was characterized as neuroparalytic. The presence of bilateral ptosis and unilateral miosis, neuroparalytic corneal ulceration and reduced pupillary light reflexes of the right eye indicated oculosympathetic paresis7,8, in the absence of space-occupying lesion of brain, neck and thorax.
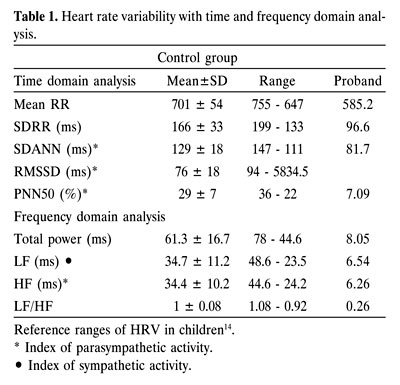
The presence of sinus tachycardia and the absence of bradycardia during severe hypoxia was suggestive of parasympathetic dysfunction6. The presence of hypoxia unawareness was also indicative of autonomic dysfunction9-11. On a 24hour electrocardiogram (ECG), sinus tachycardia was observed. Spectral analysis of heart rate variability (HRV) showed that all the patient’s HRV parameters were significantly reduced, by comparison with the age specific normal ranges12-15 (table 1).
At age 510,12 years, growth retardation was very evident (growth velocity (GV): 0.5 cm/6 months). Growth hormone (GH) levels were reduced during provocative tests (Peak GH values: Glucagon test: 3.1 µg/L, Clonidine: 2.9 µg/L, normal values> 10) and the IGF1 value was low (29 ng/ml). Prolactin levels were persistently increased: 36 to 40 £g/L. Thyroxine and TSH levels were low, (T4: 57.9 nmol/L, TSH: 1.1 mU/L) with a blunted TSH response to TRH (3.5 mU/L).
The peak cortisol level during Glucagon test was 66.2 nmol/L and the basal ACTH value was 6.4 pmol/L (1.9-11.4). Thus the diagnosis of GH and TSH deficiencies were established and the child was started on hGH and Thyroxine. After 3.5 months an adequate growth response was observed (GV: before hGH 0.5 cm/6 months and during hGH administration 3.8 cm/3.5 months, with a parallel weight gain of 1.3 kg).
Metabolic studies: The urine organic acids were normal, the plasma aminogram revealed increased glutamine value on two occasions. The significance of this finding remains unkwown.
The presence of seizures, together with behavioral problems, miosis, ptosis, temperature instability and hyperprolactinemia suggested a disorder of biogenic monoamine metabolism16. The 5HIAA (5-hydroxyindolacetic acid) value, the HVA (homovanillic acid) value and the ratio of HVA/HIAA were reduced in the CSF. Based on this presumptive diagnosis, she was administered a low dose of L-Dopa, which, however, did not result in any clinical improvement.
The episodes of apnoea/hypopnoea increased in frequency during the last months and the child remained in a hypoxic/hypercapnoic state for increasing periods of time. Oxygen was administered by mask in case of apnoeas and during sleep. A muscle biopsy for possible mitochondrial disorder was refused by the parents.
The patient died suddenly at the age of 66/12 years after an episode of prolonged cyanosis during sleep, although she was under O2 administration by mask. She was found in bed at home in the morning in a state of coma, pulseless and apnoic. She was intubated and immediately transferred to the PICU of our Hospital, where she was admitted for 24 hours. During this period she remained intubated and ventilated, under inotropic agents. During her admission she suffered two episodes of cardiac arrest. During the second episode, she failed to respond to resuscitation. Autopsy was refused by the parents.
DISCUSSION
This is a case of a 6-year old girl who, 2.5 years previous and without apparent cause, started to manifest a hypothalamo-pituitary syndrome (multiple pituitary hormone abnormalities: transient Cushing syndrome, persistent hyperprolactinemia, SIADH, GH and TSH deficiencies), together with autonomic dysfunction (episodes of apnoic spells, prolonged fever, ptosis, miosis), as well as signs of encephalopathy: seizures, myoclonias, behavioral problems.
The following etiologies could be proposed to explain the derangements described above.
1. Neurometabolic disorders (defects of the biogenic monoamine metabolites, ie. serotonin, dopamine, epinephrine and norepinephrine)16.
The characteristic CSF findings of biogenic monoamine disorders are low levels of homovanillic acid (HVA) and normal 5-hydroxyindolacetic acid (5-HIAA)16. In our patient, the CSF levels of HVA were reduced, but the 5-HIAA levels were also reduced, a finding indicative of an additional serotonin pathway dysfunction16.
2. Mitochondrial disorder
Mitochondrial disorders form a heterogeneous group of diseases with multisystem involvement. All of our patient’s symptoms, could be attributed to mitochondrial disorders17,18.
The diagnostic features of mitochondrial disorders are not well established and the characteristic changes (ragged red muscle fibers) are found in very few patients17. Serum and cerebrospinal fluid lactic acid are reported to be increased19 as well as urinary organic acids19. Our patient never had increased lactic acid in blood or CSF and no muscular problem. However, normal lactic acid concentrations do not preclude a diagnosis of mitochondrial disorder17. In fact, Nissencorn et al17 concluded that even in the absence of positive criteria, a mitochondrial disorder remains highly probable, based on clinical features.
3. Toxic or viral initiation of an autoimmune process (autoimmune hypothalamitis- hypophysitis2
Finally the relationship of our patient’s condition to her three uncle’s history of psychosis remains an open question.
ACKNOWLEDGEMENTS:
We would like to thank Dr.George Kazianis, Department of Cardiology, 7th National Insurance Funds Hospital, Athens, Greece, for his help in analyzing the 24hour ECG using power spectral analysis of heart rate variability.
REFERENCES:
1. Adrogue HJ, Madias NE, 2000 Hyponatremia. New Engl J Med 342 (21): 1581-1589.
2. Nagai Y, Ieki Y, Ohsawa K, Kobayashi K, 1997 Simultaneously found transient hypothyroidism due to Hashimoto’s thyroiditis, autoimmune hepatitis and isolated ACTH deficiency after cessation of glucocorticoid administration. Endocrinol J 44(3): 453-8.
3. Writing group of the Histiocyte Society,1987 Histiocytosis syndromes in childhood. Lancet 1: 208-210.
4. Kone-Paut I, Portas M, Wechsler B, Girard N, Raybaud C, 1999 The pitfall of silent neurosarcoidosis. Pediart Neurol 20(3): 215-218.
5. Milman N, Hoffmann AL, Byg KE, 1998 Sarcoidosis in children. Epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr 87(8): 871-878.
6. Breningstall GN, 1996 Breath-Holding Spells. Ped Neurol 14(2): 91-97.
7. Southall DP, Samuels MP, Talbert DG, 1990 Recurrent cyanotic episodes with severe arterial hypoxaemia and intrapulmonary shunting: A mechanism of sudden death. Arch Dis Child 65: 953-61.
8. Woodruff G, Buncic JR, Morin JD, 1988. Horner’s syndrome in children. J Pediatr Opthalmol and Strabism 25(1): 40-44.
9. Calverley PMA, Ewing DJ, Campbell IW, 1982 Preservation of the hypoxic drive to breathing in diabetic autonomic neuropathy. Clin Sci 63: 17-22.
10. Soler NG, Eagleton LE, 1982 Autonomic neuropathy and the ventilatory responses of diabetics to progressive hypoxaemia and hypercapnia. Diabetes 31: 609-14.
11. Ewing DJ, Clarke BF 1982 Diagnosis and management of diabetic autonomic neuropathy. Br Med J 285: 916-8.
12. Cook JR, Bigger JT, Kleiger RE, Fleiss JL, Steinman RC, Rolnitzky LM, 1991 Effect of atenolol and diltiazem on heart period variability in normal persons. J Am Coll Cardiol 17: 480-4.
13. Pomeranz B, MacAulay RJB, Caudill MA, Kutz I, Adam A, Gordon D, Kilborn KM, Barger AC, Shannon DC, Cohen RJ, Benson H, 1985 Assessment of autonomic function in humans by heart rate spectral analysis. Am J Physiol 248: M151-153.
14. Akinci A, Celiker A, Baykal E, Tezic T, 1993 Heart rate variability in diabetic children: sensitivity of time- and frequency-domain methods. Pediatr Cardiol 14: 140-6.
15. Massin M, von Bernuth G, 1997 Normal ranges of heart rate variability during infancy and childhood. Pediatr Cardiol 18: 297-302.
16. Hoffmann GF, Surtees RAH, Wevers RA, 1998 Cerebrospinal fluid investigations for neurometabolic disorders. Neuropediatrics. 29: 59-71.
17. Nissenkorn A, Zeharia A, Lev D, Fatal-Valevski A, Barash V, Gutman A, Harel S, Lerman-Sagie T, 1999 Multiple presentation of mitochondrial disorders. Arch Dis Child 81: 209-215.
18. Munnich A, Rotig A, Chretien D, Saudubray JM, Cormier V, Rustin P, 1996 Clinical presentations and laboratory investigations in respiratory chain deficiency. Eur J Pediatr 155: 262-274.
19. Trijbels JMF, Scholte HR, Ruitenbeek W, Sengers RCS, Janssen AJM, Busch HFM, 1993 Problems with the biochemical diagnosis in mitochondrial (encephalo-) myopathies. Eur J Pediatr 152: 178-184.