HORMONES 2006, 5(3):205-209
DOI: 10.14310/horm.2002.——
Address correspondence and requests for reprints to:
Konstantina Kosta, 3 Kiouptsidou Str., 55133, Kalamaria, Thessaloniki, Tel: 2310-434579, e-mail: konstantinakosta@yahoo.gr
Received 21-02-06, Revised 30-05-06, Accepted 10-06-06
Abstract
The McCune-Albright Syndrome (MAS) is a sporadic rare disease first described in 1936 by McCune and separately by Albright. MAS is characterized by a triad of physical signs: café-au-lait spots, polyostotic fibrous dysplasia and autonomous endocrine hyperfunction. MAS is predominantly observed in girls and is rarely reported in males. We report the case of a 9-year old boy with gonadotropin independent precocious puberty, café-au-lait spots, polyostotic fibrous dysplasia and growth hormone hypersecretion.
INRTODUCTION
The McCune-Albright syndrome (MAS) was first described in 1936 by McCune and separately by Albright.1,2
MAS is a sporadic disease rarely reported in males. In a cohort of 41 patients from Germany, Austria and Switzerland, only 5 patients were males,3 while in a report from France comprised of 113 patients, only 15 patients were males.4 The exact incidence of the syndrome is unknown. The diagnosis is considered confirmed when at least two of the cardinal features are present: polyostotic fibrous dysplasia, café-au-lait skin pigmentation and autonomous endocrine hyperfunction. The most common form of autonomous endocrine hyperfunction in this syndrome is gonadotropin independent precocious puberty, but affected individuals may also have autonomous hyperfunction of other endocrine glands such as hyperthyroidism,5 hypercortisolism6 and/or pituitary adenomas secreting GH and/or prolactin.7 Hypophosphatemic osteomalacia has also been reported.8 Association of MAS with hypersomatotropism is rare and pituitary adenoma is demonstrable in only 40-50% of patients with GH hypersecretion.9,10 Nonendocrine abnormalities in this syndrome include chronic liver disease, tachycardia and rarely sudden death, possibly from cardiac arrhythmias.
Herein, we report a male patient with MAS associated with GH hypersecretion.
PATIENT’S DESCRIPTION
A nine-year old boy was admitted to our department for investigation of precocious puberty. The boy presented café-au-lait spots first noted at the age of 4 months. The skin lesions were initially localised on the left shoulder and subsequently observed on the left half of the thorax, the left flank, the left upper arm and the left half of the face (Figure 1). A skin biopsy one year previously had revealed no abnormal pathology. Physical examination exposed a prominent supraorbital ridge. The height was 143cm (height SDS=2.07, 90th percentile), the weight was 40Kgr and the bone age 12.8yrs. Two and a half years previously the height was 118 cm (height SDS=0.38, 65th percentile). The mother’s height was 165cm and the father’s height 172cm, while the target height was 175cm (target height SDS=0.04). The blood pressure was 100/80mmHg. The volume of the right testis was 5ml and that of the left was 7ml. Genitalia, pubic and axillary hair were Tanner stage II. The rest of the physical examination was unremarkable as were the family and perinatal history. The birth weight was 3150gr.
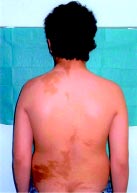
Figure 1. Café-au-lait skin pigmentation.
The basal laboratory findings are shown in Table 1 and the results of the dynamic testing are shown in Table 2 .
Skeletal radiography disclosed numerous cystic areas in the iliac and femoral bones, thickening and expansion of the base of the skull, lesions of the facial skull causing dysfigurement and scoliosis of the lumbar and thoracic spine. A Tc-99m HDP bone scintigraphy revealed abnormal concentration of the radionuclide in the iliac and femoral bones and in the facial skull (Figure 2). A thyroid ultrasonography (u/s) revealed a micronodular appearance of the gland. A testes u/s revealed microlithiasis of both testes. An abdominal u/s showed no abnormalities. Pituitary Magnetic Resonance Imaging (MRI) revealed a microadenoma with a diameter of 9mm. An adrenal MRI was normal.
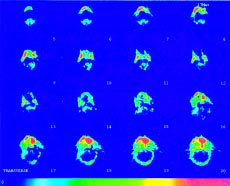
Figure 2. Tc-99m HDP bone scintigraphy. Abnormal concentration of the radionuclide in the facial skull. Fibrous dysplasia.
The diagnosis of MAS was made and treatment with somatostatin long-acting analogues (one intramuscular injection of 10mg every month) and testolactone (30mg/Kgr/day per os) was initiated. A pituitary MRI 3 years after diagnosis revealed no abnormalities. Testolactone was discontinued at the age of 13 and central puberty was initiated shortly thereafter. During a 4-year follow-up the fibrous dysplasia has not affected vision or hearing, and the GH values have been within normal range. The boy has until now presented normal physical and mental development with no other medical problem. His current height is 169cm (height SDS=1.99, 90th percentile), his weight is 71.5 Kgr, the volume of the right testis is 11.4ml and that of the left is 14ml. Pubic and axillary hair is Tanner stage III and bone age is 15.4 yrs.
DISCUSSION
MAS is the result of a postzygotic mutation in the gene encoding for the alpha subunit of the stimulatory G protein (Gsa). The mutations lead to the substitution of Histidine (His) or Cysteine (Cys) for Arginine (Arg) at aminoacid 201 of the alpha subunit of Gsa.11 The specific mutations that cause MAS occur at a site in the protein that mediates the inactivation of the Gsa subunit. Once activated, the mutated Gsa remains activated for a prolonged period despite the absence of hormone stimulation of the receptor. As a result, there are persistently high levels of intracellular cAMP. In various tissues increased cAMP levels can mediate mitogenesis and increased cell function. The specific phenotype depends on the cell type containing the mutation. The classic triad of MAS can all be explained by the mechanism described above. Since MAS results from a postzygotic mutation, the earlier the mutation occurs in embryogenesis the more widespread is tissue involvement. Mutations late in embryogenesis account for the mild cases of the syndrome with only 2 or 3 of the classic features, while mutations after differentiation into a specific cell line may lead to a single adenoma. Gsa activating mutations have been reported in isolated hyperfunctioning thyroid nodules and in somatotroph adenomas.
The most common endocrine feature of MAS is precocious puberty. It is the result of gonadotropin independent autonomous ovarian or testicular function. Precocious puberty caused by this mechanism is far more common in girls than in boys. In a cohort of 26 patients with MAS, the onset of precocious puberty was earlier in females (2.8±2.3 years) than in males (6.9±2.7).12 In our patient precocious puberty was diagnosed at the age of 8.8 years. Girls as young as 4 months with MAS may present with breast development or vaginal bleeding. Excess estrogen secretion often stimulates increased growth velocity and can result in marked advancement in skeletal maturity. Macroorchidism due to autonomous hyperfunction of Sertoli cells and Gsa mutation are also reported as an unusual expression of MAS in prepubertal boys.12 ACTH independent Cushing syndrome generally results in growth failure and hypertension in infancy. The adrenal glands are bilaterally enlarged and contain multiple nodules in the cortex. Hyperthyroidism associated with MAS is a result of one or more autonomously functioning nodules and typically occurs later in childhood. GH excess from pituitary adenomas can occur at any age resulting in gigantism and/or acromegaly. GH hypersecretion in MAS differs in several respects from that observed in classical acromegaly. Patients are generally young (<20 years) at the onset and diagnosed on the basis of growth acceleration rather than facial dysmorphism, which is usually difficult to assess due to fibrous dysplasia.9,10 Fibrous dysplasia in MAS can involve any bone but most commonly affects the long bones, the ribs and the skull. It ranges from small asymptomatic areas detectable only by bone scan to markedly dysfiguring lesions resulting in pathologic fractures and impingement on vital nerves. Fibrous dysplasia as the initial symptom of MAS has been reported in two affected boys. In our patient the lesions were located in the basal and facial skull and in the iliac and femoral bones. Hepatic abnormalities range from mild elevation of transaminases to severe neonatal jaundice and chronic cholostasis. No hepatic abnormalities were noted in our patient.
The differential diagnosis of MAS must include: gonadotropin dependent precocious puberty, neurofibromatosis, Graves’ disease, Cushing syndrome of various causes, and additionally in the male testotoxicosis.
Therapy in MAS is symptomatic. At present no therapy addresses the underlying molecular problem. Aromatase inhibitors, which block the conversion of testosterone to estradiol, are used in the treatment of precocious puberty. Pituitary surgery in this syndrome is beset with problems mainly related to fibrous dysplasia and the resultant thickening of bones. Bromocryptine, cabergoline and long-acting somatostatin analogue have been used with some success with regard to GH hypersecretion.
Our patient was presented at age 9 years with the classical triad of the syndrome: polyostotic dysplasia causing facial disfigurement, café-au-lait hyperpigmentation not crossing the midline, gonadotropin independent precocious puberty and GH hypersecretion. He received treatment with long-acting somatostatin analogues for the GH secreting adenoma and testolactone for the precocious puberty. The treatment resulted in normalization of GH secretion, complete reversion of the pituitary adenoma, no further growth acceleration and control of precocious puberty. The efficacy of the treatment applied in our patient is well established in many other reported MAS cases.13-15 Nevertheless, complete reversion of the pituitary adenoma is relatively unusual. Side effects from the treatment were not reported by the patient or his family. Despite the extended fibrous dysplasia, the boy faced no vision or hearing problems and experienced no fractures or any other kinetic problem. Four years after diagnosis the boy displays an uneventful course.
REFERENCES
1. McCune DJ, 1936 Osteitis fibrosa cystica; the case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 52: 743-744.
2. Albright F, Butler AM, Hampton AO, Smith P, 1937 Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females: report of five cases. N Engl J Med 216: 727-746.
3. Albers N, Jorgens S, Deiss D, Hauffa BP, 2002 Mc Cune-Albright syndrome–the German experience. J Pediatr Endocrinol Metab 15: Suppl 3: 897-901.
4. Lumbroso S, Paris F, Sultan C, 2004 Activating Gsalpha mutations: analysis of 113 patients with signs of Mc Cune-Albright syndrome–a European collaborative study. J Clin Endocrinol Metab 89: 2107-2113.
5. Feuillaqn PP, Shawker T, Rose SR, Jones J, Jeevanram RK, Nisula BC, 1990 Thyroid abnormalities in the McCune-Albright syndrome: ultrasonography and hormone studies. J Clin Endocrinol Metab 71: 1596-1601.
6. Mauras N, Blizzard RM, 1986 The McCune-Albright syndrome. Acta Endocrinol Suppl (Copenh) 279: 207-217.
7. Cuttler L, Jackson JA, Saeed uz-Zafar, Levitsky L, Mel-linger RC, Frohman LA, 1989 Hypersecretion of growth hormone and prolactin in McCune-Albright syndrome. J Clin Endocrinol Metab 68: 1148-1154.
8. Lee PA,Van Dop C, Migeon CJ, 1986 McCune-Albright syndrome: long-term follow-up. JAMA 256: 2980-2984.
9. Chanson P, Dib A, Visot A, Derome PJ, 1994 McCune-Albright syndrome and acromegaly: clinical studies and response to treatment in five cases. Eur J Endocrinol 131: 229-234.
10. Premawardhana LDKE, Vora JP, Mills R, Scanlon MF, 1992 Acromegaly and its treatment in the McCune-Albright syndrome. Clin Endocrinol 36: 05-608.
11. Song HD, Chen FL, Shi WJ, et al, 2002 A novel, complex heterozygous mutation within Gsalpha gene in patient with McCune-Albright syndrome. Endocrine 18: 121-128.
12. Wasniewska M, Matarazzo P, Weber G, et al, Italian Study Group for alterations of Gs alpha Protein Function, 2006 Clinical presentation of McCune-Albright syndrome in males. J Pediatr Endocrinol Metab 19: Suppl 2: 619-622.
13. Zacharin M, 2005 Paediatric management of endocrine complications in McCune-Albright syndrome. J Pediatr Endocrinol Metab 18: 33-41.
14. Schoof E, Dorr HG, Kiess W, et al, 2004 Five year follow up of a 13-year-old boy with a pituitary adenoma causing gigantism-effect of octreotide therapy. Horm Res 61: 184-189.
15. Tolis G, 1996 The role of somatostatin agonistic analogs in the treatment of acromegaly. Metabolism 45: Suppl 1: 109-110.