Abstract
Abnormalities of the Hypothalamic-Pituitary-Adrenal (HPA) axis have been documented in HIV patients in the early as well as late stages of the infection and range from subtle subclinical disturbances to frank adrenal insufficiency. Potential etiologies of these disorders include opportunistic infections, neoplasms, drugs administered to treat infections, cytokine abnormalities associated with the HIV disease process and acquired alterations in tissue sensitivity to glucocorticoids. In this article, we present a concise review of HPA abnormalities in HIV infection and disease with regard to their etiology with emphasis on syndromes of hypersensitivity/resistance to glucocorticoids associated with antiviral medications and/or the HIV infection itself.
HPA axis
The HPA axis and the systemic sympathetic/adrenomedullary (sympathoadrenal) system are the peripheral branches of the stress system, whose main function is to maintain basal and stress-related homeostasis.1 Biologic, physical or psychologic stimuli activate the stress system, including the HPA axis. Such activation has been referred to as stress response.2 The primary mediator of this response is corticotropin-releasing hormone (CRH), a 41-amino acid peptide that plays a central role in coordinating the HPA axis and the systemic response to stress,3 acting as the main physiologic ACTH stimulus.4 ACTH leads to secretion of cortisol (F) and other adrenal steroids, such as dehydroepiandrosterone (DHEA) and aldosterone.5
The HPA axis and the immune-inflammatory response
Complex interactions exist between the HPA axis and the immune system.1,6,7 Several inflammatory signals, mainly consisting of circulating cytokines, are produced as a result of innate and adaptive immune system activation. These cytokines play a major role in the protection of the organism from foreign attacks and in the pathogenesis of autoimmune processes,8 accounting for most of HPA axis activation during an ongoing infectious or inflammatory disease. Among them, tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1 and IL-6 activate the HPA axis independently, albeit in a synergistic way.9
In addition to their effects on the hypothalamus, cytokines can stimulate ACTH and cortisol release by acting directly on the pituitary and adrenals, respectively.10,11 Other inflammatory mediators, like interferon-α, interferon-γ, interleukin-2, epidermal growth factor (EGF), transforming growth factor-β (TGF-β) and platelet activating factor (PAF), may also participate in the regulation of the HPA axis, directly or indirectly, by stimulating the release of inflammatory cytokines.1,3
Activation of the HPA axis and hence the release of glucocorticoids has a pivotal role in adaptation during the stress of infection by modulating the immune inflammatory response. Glucocorticoids suppress immune activation of inflammatory cells, inhibit the production of cytokines (TNF-α, IL-1β, IL-6) and other inflammatory mediators1,3 and suppress certain subgroups of lymphocytes, namely Th1 lymphocytes.1,3 Certain cytokines (IL-2, IL-4) can induce resistance to glucocorticoids by decreasing the affinity of the glucocorticoid receptor to its ligand.12 In HIV infection and disease, cytokines have a central role in activating the HPA axis and thus in the regulation of the immune response (Figure 1).
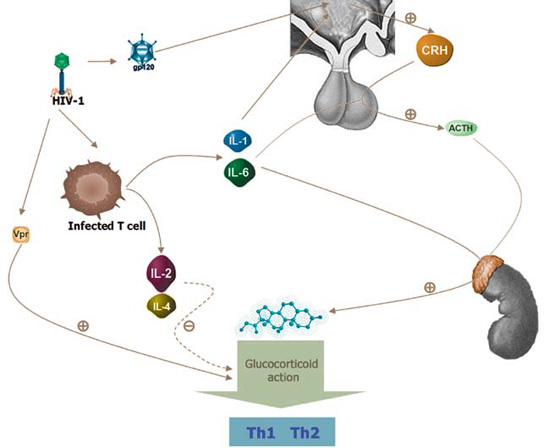
Figure 1. Schematic representation of HIV-1 effects on the HPA axis. HIV-1 stimulates glucocorticoid action by increasing glucocorticoid sensitivity through the direct effect of Vpr in target tissues. HIV-1 envelope protein gp120 induces glucocorticoid secretion by direct stimulation of the HPA axis. Interleukins-1 and -6 have a stimulatory effect on glucocorticoid secretion both directly by inducing secretion via the adrenal cortex and indirectly by activating CRH and ACTH secretion. A specific subset of patients exhibits glucocorticoid resistant-like phenotype probably attributable to the effects of interleukins IL-2 and IL-4 secreted by infected T cells on glucocorticoid action.
HPA FUNCTION IN HIV PATIENTS
The HPA axis has been examined extensively in AIDS and HIV-infected patients. High13,14 or normal15,16 basal cortisol levels and high,16,17 low17,18 or normal19 ACTH plasma levels have been reported. Evaluation using CRH and/or ACTH has revealed derangement of the HPA axis ranging from subclinical alterations in cortisol levels to frank adrenal insufficiency, depending on whether the subjects were patients in the early or advanced stages of HIV infection.
In patients with early HIV infection, adrenal stimulation with ACTH (cosyntropin test) revealed subnormal responses in 8-14% of cases,16,18,19 whereas in advanced HIV infection and in AIDS the respective rate was 54%.19 Patients with elevated baseline levels of cortisol associated with blunted responsiveness to cosyntropin were also reported.20 The significance of such blunted responses is not clear. Evaluation of the HPA axis with CRH test in patients with early HIV infection who had normal responses to cosyntropin revealed reduced cortisol responsiveness,19,20 suggesting that many asymptomatic patients in the early stages of the disease may have subclinical primary or secondary adrenal insufficiency. The low responsiveness of the pituitary to CRH was attributed to a reversible effect of cytokines.13 In a study of 25 asymptomatic HIV patients, normal adrenal capacity was reported; however, 32% had elevated plasma ACTH at the two-year follow-up, suggesting a progressively compromised adrenal reserve.21 With progression towards AIDS, overt adrenal insufficiency may be established in some patients, often secondary. However, in AIDS patients, adrenal function is rarely compromised and the adrenal insufficiency observed in some of them can be attributed to specific causes, such as adrenal or pituitary infection or neoplastic infiltration, and is associated with the typical biochemical features of Addison’s disease.
PATHOGENETIC MECHANISMS OF HPA AXIS DYSFUNCTION IN HIV PATIENTS
Hypercortisolemia in AIDS patients
In a considerable number of AIDS patients, elevated basal cortisol levels have been observed.19 This might be due to a chronic stress-related shift of steroid production from adrenal androgens toward cortisol. Another possible explanation is the increasing plasma concentrations of cortisol-binding globulin (CBG) observed with the progression of the disease.22 High cortisol levels associated with low ACTH levels may indicate that in HIV infection, cytokines, such as IL-1β and IL-6, directly stimulate the adrenal glucocorticoid synthesis pathway.13,18 Concomitant high levels of ACTH and cortisol often observed in these patients suggest a stimulatory effect of these cytokines on CRH release.23 In addition, more recent data suggest that the HIV envelope protein gp-120 might induce HPA axis hyperactivity.25 Whether the observed high cortisol levels are beneficial, due to the anti-inflammatory properties of this hormone, or deleterious, as a result of its immunosuppressive properties, remains unclear. However, as in other forms of acute or chronic illness, it might reflect an adaptive, albeit allostatic, stress response.
Glucocorticoid resistance:
A subset of AIDS patients have clinical manifestations indicative of adrenal insufficiency (hypotension, fatigue, skin and mucosal pigmentation) in parallel with a biochemically hyperfunctioning HPA axis characterized by hypercortisolemia and a moderate increase of ACTH.26 In these patients, glucocorticoid affinity to its ligand was reduced and the glucocorticoid receptor (GR) number was increased, suggesting a partial glucocorticoid resistance state. As mentioned above, this phenomenon might be due to altered cytokine action. A similar glucocorticoid resistance state is present in glucocorticoid resistant asthma type 2 patients. These patients have a specific cytokine pattern consisting mainly of elevated IL-2 and IL-4 and also exhibit reduced glucocorticoid affinity to its ligand, closely resembling the glucocorticoid resistance state found in the aforementioned AIDS patients.27 Another possible explanation for the relative glucocorticoid resistance of these patients is the increased expression of the GRβ splicing variant of the glucocorticoid receptor relatively to the expression of the GRα isoform.28 The GRα isoform is the main mediator of glucocorticoid activity and the GRβ isoform is known to inhibit GRα action. Thus, the increased intracellular proportion of GRβ isoform leads to a decreased glucocorticoid effect.29,30
Involvement of other adrenal zones
Mineralocorticoids:
Both hyperreninemic and hyporeninemic hypoaldosteronism have been found in HIV-infected patients.31 Although frank mineralocorticoid deficiency is uncommon, an impaired aldosterone response to ACTH is observed in many HIV-infected patients.21 Hyponatremia and hyperkalemia are also common findings in AIDS patients. However, these abnormalities are most likely due to drug toxicities rather than the HIV infection itself. Other causes of electrolyte abnormalities include interstitial kidney disease and the syndrome of inappropriate ADH secretion secondary to pulmonary or central nervous system processes.32 Two cases of primary hyperaldosteronism have been reported in HIV-infected patients.33 Ιt was proposed that hyperaldosteronism in these cases might be due to the intrinsic renin-like activity of the HIV aspartic protease.
Adrenal androgens:
In HIV-infected patients, steroid metabolism exhibits a shift from adrenal androgens and 17-deoxysteroids towards cortisol.34 A similar pattern of shunting away from adrenal androgens and towards cortisol has been observed in acute and chronic illness, as well as in malnutrition among non-HIV patients.35 Such a pattern of high cortisol and reduced DHEA may be due to a decreased adrenal 17,20-lyase activity.36 In a study investigating ovarian and adrenal function in HIV-infected women with AIDS, significantly reduced DHEA and increased cortisol responses to ACTH were demonstrated. The ratio of the DHEA to cortisol response (an index of shunting away from adrenal androgen secretion and towards increased cortisol production) was significantly decreased in women with AIDS-related wasting syndrome compared to control subjects. In contrast, the same study demonstrated an intact ovarian androgen responsiveness to hCG stimulation.37,38
Influence of altered steroid production on immune status in AIDS
Plasma DHEA concentrations correlate positively with the CD4 cell count.39 Also, certain studies demonstrate a negative linear correlation between CD4 count and cortisol levels,40 while others do not.39 It has been suggested that the pattern of high cortisol/low DHEA-S levels is associated with HIV illness markers, including viral load, and that this finding carries a negative prognostic value.40 In fact, a reduced DHEA-S/cortisol ratio is associated with a deterioration of the immune status of HIV-infected patients due to a shift from a Th1- to a Th2-driven immune response.41 Thus, AIDS patients with diminished DHEA-S levels display an excessive production of cytokines by Th2 cells (IL-4, IL-5, IL-6, IL-10) and a suppression of cytokines (IL-2, IFN-γ, IL-12) secreted by Th1 cells, the latter apparently negatively affecting the clinical course of the disease. DHEA-S supplementation appears to restore cytokine production in animal models.42
ADRENAL INSUFFICIENCY IN HIV-INFECTED PATIENTS
Etiology:
Concomitant opportunistic infections in patients with HIV infection may compromise adrenal function by acting at several levels of the HPA axis (Table 1 ). Adrenal or pituitary infection can lead to adrenal insufficiency which can be subclinical or overt with the progression towards AIDS.43 Direct HIV infection of the adrenals and adrenal neoplasms (Kaposi’s sarcoma, lymphoma) can impair adrenal function directly. Anti-adrenal antibodies have been detected in HIV patients, probably reflecting thymic dysfunction or an epiphenomenon linked to nonspecific B-cell activation.44 Several medications commonly used in the treatment of HIV-infected patients interfere with adrenal function. Among these, ketoconazole inhibits steroidogenesis, while rifampin and phenytoin enhance steroid metabolism and may unmask adrenal insufficiency in patients with a decreased adrenal reserve.45-47 The progestational agent megestrol acetate exhibits intrinsic glucocorticoid activity and prolonged administration can induce secondary adrenal insufficiency. Abrupt withdrawal of megestrol acetate, especially after chronic use, can precipitate acute adrenal insufficiency48 (Table 1 ).
Autopsy findings:
Autopsy studies have revealed adrenal gland involvement in 40-90% of cases and pituitary gland involvement in 30% of cases.49-51 Adrenal and pituitary functions may be affected by infection, malignancy, hemorrhage, necrosis and fibrosis. Cytomegalovirus infection of the adrenals is the most common finding at autopsies. CMV adrenalitis is characterized by the presence of intracytoplasmic and intranuclear inclusion bodies in enlarged adrenal glands.52 Adrenal insufficiency arises after more than 80% of adrenal tissue has been destroyed.53 Interestingly, 3% of the autopsies performed in unselected patients with AIDS revealed CMV infection and adrenal necrosis in more than 80% of adrenal tissue.54 This finding is consistent with that derived from another autopsy series, in which the frequency of adrenal insufficiency in antemortem studies was 3%.55 Opportunistic infections by Mycobacterium avium-complex (MAC), Mycobacterium tuberculosis, Cryptococcus neoformans, Toxoplasma gondii, Pneumocystis carinii and Histoplasma capsulatum have also been found on pathologic examination of the adrenals.56
MANAGEMENT OF ADRENAL INSUFFICIENCY IN HIV-INFECTED PATIENTS
Identification of adrenal insufficiency in HIV-infected patients is imperative because treatment with corticosteroids might be life-saving. On the other hand, institution of therapy without confirmed adrenal insufficiency might worsen underlying opportunistic infections. The diagnosis of adrenal insufficiency in the setting of HIV infection may be challenging because many of these patients have nonspecific symptoms such as fatigue, weight loss, nausea and vomiting, resembling those of adrenal insufficiency. However, prompt evaluation of HPA axis function should be performed in all end-stage AIDS patients, in patients with specific manifestations of adrenal insufficiency (skin and mucosa hyperpigmentation, hyponatremia and hyperkalemia) and in patients at increased risk of developing adrenal insufficiency (patients with tuberculosis or disseminated cytomegalovirus infection). In such patients, determination of baseline plasma cortisol and ACTH levels and a cosyntropin stimulation test should be carried out. As in other forms of adrenal insufficiency, if the response to cosyntropin is inadequate, patients should be treated for adrenal insufficiency. If the response to the cosyntropin test is normal and there is still a strong suspicion of secondary adrenal insufficiency (for example, due to hypopituitarism, glucocorticoid or megestrol acetate use), then testing with CRH or insulin-induced hypoglycemia should be performed for the evaluation of the integrity of the HPA axis.
HIV patients with increased baseline cortisol levels might have blunted response to the cosyntropin test, suggesting impaired adrenal reserve. The management of this subset of patients is often challenging. Among this subset, hydrocortisone supplementation should be administered cautiously because chronic glucocorticoid therapy may have significant adverse consequences in these individuals, who are already immunocompromised.57 Whenever adrenal insufficiency can be attributed to an identifiable etiologic factor, specific therapeutic measures should be undertaken. If drugs known to impair adrenal function are being administered to AIDS patients, then alternative therapeutic methods should be sought, using agents known not to affect the HPA axis, whenever possible. For example, fluconazole, which does not appear to affect adrenal function, may be used instead of ketoconazole. The action of some agents is reversible. For example, hypocortisolism induced by rifampin has been shown46 to subside after drug withdrawal. In cases in which the offending agent cannot be substituted or discontinued, evaluation of HPA axis function should be performed, and in patients with abnormal adrenal function, hydrocortisone replacement therapy should be instituted.
Patients with documented adrenal insufficiency should be treated with hydrocortisone 15-25 mg per day in two divided doses, with the larger dose administered in the morning. In patients with primary adrenal insufficiency, the addition of mineralocorticoid replacement might be required (fludrocortisone acetate 0.05-0.2 mg per day). As in any case of adrenal insufficiency, the dose of hydrocortisone should be increased up to 2 to 3 times in moderate stress states. In cases of severe stress, patients will require even higher doses of glucocorticoids (up to 300 mg hydrocortisone per day). Increased doses of hydrocortisone may also be required in cases of concomitant administration of agents known to stimulate cortisol clearance such as phenyntoin. If rifampin is used (i.e. for extrapulmonary tuberculosis treatment), then doubling or tripling the dose of adrenal steroids is recommended46
AIDS-RELATED INSULIN RESISTANCE AND LIPODYSTROPHY SYNDROMES
Extensive use of antiretroviral drugs and protease inhibitors yields significantly improved survival rates of AIDS patients. However, this has led to the emergence of novel morbidities that pose an important, albeit less imminent threat for these patients. Central among them is the AIDS-related insulin resistance and lipodystrophy syndrome (ARIRLS).58-60 The risk factors of this syndrome include: older age (older than 40 years of age), female sex, elevated serum triglyceride levels, low nadir CD4 cell count and an advanced stage of HIV infection.61 The phenotype of the syndrome consists of central obesity, adipose tissue redistribution (visceral fat accumulation), a `buffalo hump’, extremity thinning and muscle wasting, marked dyslipidemia and insulin resistance. Since the clinical manifestations of this syndrome closely resemble those of chronic glucocorticoid excess as seen in Cushing’s syndrome, the term ‘pseudo-Cushing’ was initially used to describe this syndrome.62 However, HPA axis hyperactivity, which is commonly seen in HIV-infected patients, seems to be an unlikely cause of ARIRLS. These patients have normal baseline (and CRH-stimulated) ACTH and cortisol values concomitantly with normal excretion of 24-h urinary free cortisol.63 Cytokine abnormalities commonly seen in AIDS patients, such as the already mentioned increase in TNF-α, IL-1β and IL-6, may also contribute to the insulin resistance observed in these patients, either by acting directly on the insulin signaling system or by stimulating the target tissue 11-beta-oxidoreductase of cortisone, converting it to cortisol in target tissue.62 Other explanations include antiretroviral regiments and protease inhibitors, in particular, as well as various viral agents as indicated below.
Protease inhibitors (PIs)
AIDS-related lipodystrophy was initially reported in patients under treatment with protease inhibitors. Thus, the pathogenesis of the syndrome was assumed to be related to an adverse effect of these drugs. Protease inhibitors alter lipid and carbohydrate metabolism,64 induce adipocyte apoptosis (sparing visceral adipose tissue),65 provoke mitochondrial injury,66 reduce glucose uptake and phosphorylation by skeletal muscles,67 cause cytokine abnormalities,53 lower adiponectin levels68 and block lamin A synthesis (thus leading to accumulation of prelamin A in adipose tissue).69 Of particular interest is the fact that HIV-1 protease has about 60% homology with the low-density lipoprotein receptor-like protein (LRP) and cytoplasmic retinoic-acid binding protein type 1 (CRABP-1).62,63 LRP plays an important role in hepatocyte and adipocyte triglyceride metabolism and protease inhibitors lower LRP levels.70 A critical step of the protease inhibitor-induced lipodystrophy is the inhibition of adipocyte expression of PPAR-γ, which might induce apoptosis and impaired differentiation of peripheral adipocytes, resulting in fat redistribution.71 Thiazolidinediones (PPAR-γ agonists) increase insulin sensitivity in HIV lipoatrophic patients.72 Furthermore, treatment with leptin at physiologic doses in a subset of HIV-infected, leptin-deficient patients improves insulin resistance and truncal fat mass and appears to represent a viable therapeutic option for this syndrome.73 However, there is still no curative treatment for the morphological features of lipodystrophy. Interruption of PIs treatment may ameliorate dyslipidemia and insulin resistance, but not the morphological changes.61 Switching treatment from PIs to other agents (nucleoside reverse transcriptase inhibitors, -non-NRTIs-, abacavir), has been suggested to improve the morphological abnormalities of lipodystrophy syndrome.61 Finally, novel protease inhibitors not affecting the ARIRLS-related pathways are under development (such as darunavir that has been shown not to interfere with the biosynthesis of lamin A, a key molecule in congenital lipodystrophies).74
Viral factors:
There is substantial evidence indicating that AIDS patients have tissue-specific glucocorticoid hypersensitivity. The cytokine profile seen in these patients (decrease in IL-2, IL-12 and IFN-γ concomitant with an increase in IL-4 levels75,76) reflects the reduction in innate and adaptive immunity and is also seen in situations characterized by significant glucocorticoid excess. In addition, muscle wasting and/or myopathy, dyslipidemia and visceral obesity-associated insulin resistance (either isolated or as part of ARIRLS) are frequent complications of HIV infection77-79 and are conditions suggestive of a possible chronic hypercortisolemic state. Since HPA axis hyperactivity (a common finding in HIV-infected patients) seems to be an unlikely cause of ARIRLS, it has been hypothesized that an increase in glucocorticoid sensitivity might, at least partially, explain some of these alterations.80,81
Much attention has been focused on HIV-1-encoded accessory proteins Vpr and Tat as possible candidates responsible for the noted glucocorticoid hypersensitivity. Vpr is a 96-amino acid virion-associated accessory protein with multiple functions,82 including host cell arrest in the G2/M phase of the cell cycle (through interaction with novel 14-3-3 proteins),82-85 transcription activation of several viral promoters,85 nuclear translocation, induction of apoptosis86 and enhancement of HIV-1 long terminal repeat (LTR) promoter (important for the expression of HIV-1 encoded proteins87) activation by Tat.85 Vpr may also exhibit paracrine and endocrine functions in distant non-infected tissues.88,89 Vpr increases glucocorticoid sensitivity in target tissues. The possible mechanisms involving coactivator or corepressor Vpr action are described below:
- The conserved LXXLL motif sequence located at amino acids 64 to 68 of the Vpr molecule is known to directly bind to the glucocorticoid receptor (GR) and is considered essential in nuclear receptor and coactivator molecule interaction.90 This sequence is found in several copies of various well-known coactivator molecules (p300/CBP and members of the p160 family of coactivators).91
- Vpr acts as an adaptor molecule among promoter bound transcriptional factors and CREB-binding protein (CBP), a well-known coactivator molecule.92
- When Vpr was administered experimentally in peripheral monocytes, IL-12 production, through augmentation of GR activity, was found to be markedly decreased.93
- Vpr has been shown to directly inhibit peroxisome proliferation activating receptor γ (PPAR-γ) action which is considered critical for the development and proper function of adipose tissue.94
- Vpr inhibits insulin induced translocation into the cytoplasm and subsequent inactivation of Foxo3a (a member of the FOXO family of proteins playing an important role in the transactivation of genes related to gluconeogenesis and glucolysis).82
Tat is considered the most potent transactivator protein of the HIV-1 LTR promoter. Similarly to Vpr, it circulates in blood and may affect target cells regardless of whether they are infected or not.95 It exerts its glucocorticoid-enhancing actions by virtue of its direct interaction with coactivator molecules (p300/CBP and p160)92 and by helping accumulate the positive transcription elongation factor-b (pTEF-b) on glucocorticoid-responsive promoters.96
In summary, ARIRLS seems to be the result of the effect of various viral factors in conjunction with the use of protease inhibitors. Prior to treatment with these agents, AIDS-related cachexia might mask the expression of insulin resistance and lipodystrophy phenotype. After treatment initiation, however, the significant clinical improvement seen in these patients (including weight regain), the direct effect of protease inhibitors, along with the persistent Vpr- and Tat-induced metabolic abnormalities allow the clinical manifestations of the typical ARIRLS to become apparent (Figure 2).
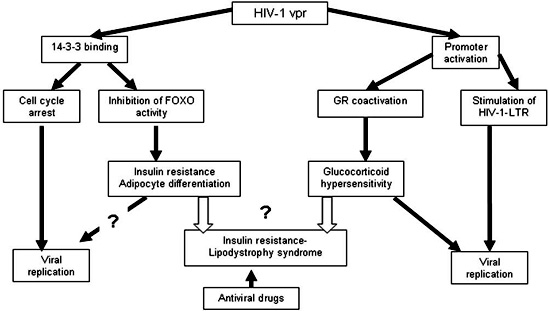
Figure 2. Possible pathogenetic mechanisms (viral factors and protease inhibitors) leading to AIDS-related lipodystrophy/insulin resistance syndrome.82
CONCLUSIONS
HPA axis dysfunction is usually encountered in HIV-infected patients. The most common dysfunction in these patients is hypercortisolemia, which is also observed in other forms of acute or chronic illnesses and is probably due to elevation of circulating cytokines. The release of cytokines, such as IL-1β and IL-6, may induce the production of glucocorticoids indirectly (via the release of CRH and/or ACTH) or directly by stimulating adrenal glucocorticoid biosynthesis. However, hypercortisolemia is a more common feature of early stage HIV infection. As the underlying disease progresses and the patients enter a more immunocompromised state, the likelihood of adrenal insufficiency gradually increases. Adrenal insufficiency is usually the result of opportunistic infections (mainly CMV) or neoplastic infiltration of the adrenals and/or the pituitary. Drugs commonly used in AIDS patients may also be a causative or triggering factor leading to adrenal insufficiency. When adrenal insufficiency is documented, prompt therapy must be instituted. However, frank adrenal insufficiency is relatively rare. Thus, provocative tests should be performed prior to treatment because glucocorticoid treatment might be harmful to AIDS patients whose immune system is already compromised.
In summary, HIV-1 per se affects the HPA axis in several ways. It induces glucocorticoid secretion through direct stimulation of the HPA axis by the HIV-1 envelope protein gp120. Conversely, it may impair adrenal function by direct infection of the adrenals. Finally, through the Vpr and Tat, HIV-1-encoded proteins, the virus may stimulate host target tissue glucocorticoid sensitivity in some AIDS patients, resulting in a clinical syndrome with the morphologic and biochemical manifestations of glucocorticoid excess (AIDS-related insulin resistance and lipodystrophy syndrome, -ARIRLS-). The etiology of this syndrome, however, seems to be multifactorial. In addition to the HIV infection itself, antiviral drugs and hypercytokinemia may contribute to the pathologic manifestations of ARIRL syndrome.
REFERENCES
1. Chrousos GP, 1995 The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332: 1351-1362.
2. Selye H, 1936 A Syndrome Produced by Diverse Nocuous Agents. Nature 138: 32.
3. Chrousos GP, Gold PW, 1992 The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 267: 1244-1252.
4. Rivier C, Brownstein M, Spiess J, Rivier J, Vale W, 1982 In vivo corticotropin-releasing factor-induced secretion of adrenocorticotropin, beta-endorphin, and corticosterone. Endocrinology 110: 272-278.
5. Conaglen JV, Donald RA, Espiner EA, Livesey JH, Nicholls MG, 1984 The effect of ovine corticotropin-releasing factor on catecholamine, vasopressin, and aldosterone secretion in normal man. J Clin Endocrinol Metab 58: 463-466.
6. Mastorakos G, Magiakou MA, Chrousos GP, 1995 Effects of the immune/inflammatory reaction on the hypothalamic-pituitary-adrenal axis. Ann N Y Acad Sci 1771: 438-448.
7. Mastorakos G, Karoutsou EI, Mizamtsidi M, 2006 Corticotropin releasing hormone and the immune/inflammatory response Eur J Endocrinol 155: Suppl 1: S77-S84
8. Mastorakos G, Ilias I, 2000 Relationship between interleukin-6 (IL-6) and hypothalamic-pituitary-adrenal axis hormones in rheumatoid arthritis. Z Rheumatol 59: Suppl 2: 75-79.
9. Imura H, Fukata J, Mori T, 1991 Cytokines and endocrine function: an interaction between the immune and neuroendocrine systems. Clin Endocrinol (Oxf) 35: 107-115.
10. Mastorakos G, Chrousos GP, Weber JS, 1993 Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab 77: 1690-1694.
11. Mastorakos G, Weber JS, Magiakou MA, Gunn H, Chrousos GP, 1994 Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. J Clin Endocrinol Metab 79: 934-939.
12. Kam JC, Szefler SJ, Surs W, Sher ER, Leung DY, 1993 Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol 151: 3460-3466.
13. Biglino A, Limone P, Forno B et al, 1995 Altered adrenocorticotropin and cortisol response to corticotropin-releasing hormone in HIV-1 infection. Eur J Endocrinol 133: 173-179.
14. Hilton CW, Harrington PT, Prasad C, Svec F, 1988 Adrenal insufficiency in the acquired immunodeficiency syndrome. South Med J 81: 1493-1495.
15. Raffi F, Brisseau JM, Planchon B, Remi JP, Barrier JH, Grolleau JY, 1991 Endocrine function in 98 HIV-infected patients: a prospective study. AIDS 5: 729-733.
16. Dobs AS, Dempsey MA, Ladenson PW, Polk BF, 1988 Endocrine disorders in men infected with human immunodeficiency virus. Am J Med 84: 611-616.
17. Verges B, Chavanet P, Desgres J et al, 1989 Adrenal function in HIV infected patients. Acta Endocrinol (Copenh) 121: 633-637.
18. Villette JM, Bourin P, Doinel C, et al, 1990 Circadian variations in plasma levels of hypophyseal, adrenocortical and testicular hormones in men infected with human immunodeficiency virus. J Clin Endocrinol Metab 70: 572-577.
19. Membreno L, Irony I, Dere W, Klein R, Biglieri EG, Cobb E, 1987 Adrenocortical function in acquired immunodeficiency syndrome. J Clin Endocrinol Metab 65: 482-487.
20. Freda PU, Papadopoulos AD, Wardlaw SL, Goland RS, 1997 Spectrum of Adrenal Dysfunction in Patients with Acquired Immunodeficiency Syndrome – Evaluation of Adrenal and Pituitary Reserve with ACTH and Corticotropin-Releasing Hormone Testing. Trends Endocrinol Metab 8: 173-180.
21. Findling JW, Buggy BP, Gilson IH, Brummitt CF, Bernstein BM, Raff H, 1994 Longitudinal evaluation of adrenocortical function in patients infected with the human immunodeficiency virus. J Clin Endocrinol Metab 79: 1091-1096.
22. Schurmeyer TH, Muller V, von Zur MA, Schmidt RE, 1997 Thyroid and adrenal function in HIV-infected outpatients. Eur J Med Res 2: 220-226.
23. Raber J, Sorg O, Horn TF, et al, 1998 Inflammatory cytokines: putative regulators of neuronal and neuro-endocrine function. Brain Res Brain Res Rev 26: 320-326.
24. Hashemi FB, Hughes TK, Smith EM, 1998 Human immunodeficiency virus induction of corticotropin in lymphoid cells. J Clin Endocrinol Metab 83: 4373-4381.
25. Costa A, Nappi RE, Polatti F, Poma A, Grossman AB, Nappi G, 2000 Stimulating effect of HIV-1 coat protein gp120 on corticotropin-releasing hormone and arginine vasopressin in the rat hypothalamus: involvement of nitric oxide. Exp Neurol 166: 376-384.
26. Norbiato G, Bevilacqua M, Vago T, et al, 1992 Cortisol resistance in acquired immunodeficiency syndrome. J Clin Endocrinol Metab 74: 608-613.
27. Leung DY, Martin RJ, Szefler SJ, et al, 1995 Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J Exp Med 181: 33-40.
28. Leung DY, Hamid Q, Vottero A, et al, 1997 Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med 186: 1567-1574.
29. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP, 1995 Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95: 2435-2441.
30. Charmandari E, Chrousos GP, Ichijo T, et al, 2005 The human glucocorticoid receptor (hGR) beta isoform suppresses the transcriptional activity of hGRalpha by interfering with formation of active coactivator complexes. Mol Endocrinol 19: 52-64.
31. Seney FD Jr, Burns DK, Silva FG, 1990 Acquired immunodeficiency syndrome and the kidney. Am J Kidney Dis 16: 1-13.
32. Sellmeyer DE, Grunfeld C, 1996 Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immune deficiency syndrome. Endocr Rev 17: 518-532.
33. Stricker RB, Goldberg DA, Hu C, Hsu JW, Goldberg B, 1999 A syndrome resembling primary aldosteronism (Conn syndrome) in untreated HIV disease. AIDS 13: 1791-1792.
34. Grinspoon SK, Bilezikian JP, 1992 HIV disease and the endocrine system. N Engl J Med 327: 1360-1365.
35. Parker LN, Levin ER, Lifrak ET, 1985 Evidence for adrenocortical adaptation to severe illness. J Clin Endocrinol Metab 60: 947-952.
36. Zumoff B, Walsh BT, Katz JL, et al, 1983 Subnormal plasma dehydroisoandrosterone to cortisol ratio in anorexia nervosa: a second hormonal parameter of ontogenic regression. J Clin Endocrinol Metab 56: 668-672.
37. Grinspoon S, Corcoran C, Stanley T, Rabe J, Wilkie S, 2001 Mechanisms of androgen deficiency in human immunodeficiency virus-infected women with the wasting syndrome. J Clin Endocrinol Metab 86: 4120-4126.
38. Zumoff B, Walsh BT, Katz JL, et al, 1983 Subnormal plasma dehydroisoandrosterone to cortisol ratio in anorexia nervosa: a second hormonal parameter of ontogenic regression. J Clin Endocrinol Metab 56: 668-672.
39. Wisniewski TL, Hilton CW, Morse EV, Svec F, 1993 The relationship of serum DHEA-S and cortisol levels to measures of immune function in human immunodeficiency virus-related illness. Am J Med Sci 305: 79-83.
40. Christeff N, Gherbi N, Mammes O, et al, 1997 Serum cortisol and DHEA concentrations during HIV infection. Psychoneuroendocrinology 22: Suppl 1: 11-18.
41. Clerici M, Trabattoni D, Piconi S, et al, 1997 A possible role for the cortisol/anticortisols imbalance in the progression of human immunodeficiency virus. Psychoneuroendocrinology 22: Suppl 1: 27-31
42. Araneo BA, Woods ML, Daynes RA, 1993 Reversal of the immunosenescent phenotype by dehydroepiandrosterone: hormone treatment provides an adjuvant effect on the immunization of aged mice with recombinant hepatitis B surface antigen. J Infect Dis 167: 830-840.
43. Eledrisi MS, Verghese AC, 2001 Adrenal insufficiency in HIV infection: a review and recommendations. Am J Med Sci 321: 137-144.
44. Salim YS, Faber V, Wiik A, Andersen PL, Hoier-Madsen M, Mouritsen S, 1988 Anti-corticosteroid antibodies in AIDS patients. APMIS 96: 889-894.
45. Putignano P, Kaltsas GA, Satta MA, Grossman AB, 1998 The effects of anti-convulsant drugs on adrenal function. Horm Metab Res 30: 389-397.
46. Kyriazopoulou V, Parparousi O, Vagenakis AG, 1984 Rifampicin-induced adrenal crisis in addisonian patients receiving corticosteroid replacement therapy. J Clin Endocrinol Metab 59: 1204-1206.
47. Ediger SK, Isley WL, 1988 Rifampicin-induced adrenal insufficiency in the acquired immunodeficiency syndrome: difficulties in diagnosis and treatment. Postgrad Med J 64: 405-406.
48. Leinung MC, Liporace R, Miller CH, 1995 Induction of adrenal suppression by megestrol acetate in patients with AIDS Ann Intern Med 122: 843-845.
49. Niedt GW, Schinella RA, 1985 Acquired immunodeficiency syndrome. Clinicopathologic study of 56 autopsies. Arch Pathol Lab Med 109: 727-734.
50. Tapper ML, Rotterdam HZ, Lerner CW, Al’Khafaji K, Seitzman PA, 1984 Adrenal necrosis in the acquired immunodeficiency syndrome. Ann Intern Med 100: 239-241.
51. Welch K, Finkbeiner W, Alpers CE, et al, 1984 Autopsy findings in the acquired immune deficiency syndrome. JAMA 252: 1152-1159.
52. Masharani U, Schambelan M, 1993 The endocrine complications of acquired immunodeficiency syndrome. Adv Intern Med 38: 323-336.
53. Mayo J, Collazos J, Martinez E, Ibarra S, 2002 Adrenal Function in the Human Immunodeficiency Virus-Infected Patient. Arch Intern Med 162: 1095-1098.
54. McKenzie R, Travis WD, Dolan SA, et al, 1991 The causes of death in patients with human immunodeficiency virus infection: a clinical and pathologic study with emphasis on the role of pulmonary diseases. Medicine (Baltimore) 70: 326-343.
55. Bricaire F, Marche C, Zoubi D, Regnier B, Saimot AG, 1988 Adrenocortical lesions and AIDS. Lancet 1: 881.
56. Hofbauer LC, Heufelder AE, 1996 Endocrine implications of human immunodeficiency virus infection. Medicine (Baltimore) 75: 262-278.
57. Freda PU, Bilezikian JP, 1999 The hypothalamus-pituitary-adrenal axis in HIV disease. AIDS Read 9: 43-50.
58. Koutkia P, Grinspoon S, 2004 HIV-Associated Lipodystrophy: Pathogenesis, Prognosis, Treatment, and Controversies. Annu Rev Med 55: 303-317.
59. Martinez E, Mocroft A, Garcia-Viejo MA, et al, 2001 Risk of lipodystrophy in HIV-1-infected patients treated with protease inhibitors: a prospective cohort study. Lancet 357: 592-598.
60. Carr A, Samaras K, Burton S, et al, 1998 A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 12: F51-F58.
61 Baril JG, Junod P, Leblanc R, et al, 2005 HIV-associated lipodystrophy syndrome: A review of clinical aspects. Can J Infect Dis Med Microbiol 16: 233-243.
62. Miller KK, Daly PA, Sentochnik D, 1998 Pseudo-Cushing’s syndrome in human immunodeficiency virus-infected patients. Clin Infect Dis 27: 68.
63. Yanovski JA, Miller KD, Kino T, et al, 1999 Endocrine and metabolic evaluation of human immunodeficiency virus-infected patients with evidence of protease inhibitor-associated lipodystrophy. J Clin Endocrinol Metab 84: 1925-1931.
64. Carr A, Samaras K, Chisholm DJ, Cooper DA, 1998 Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 351: 1881-1883.
65. Chen D, Misra A, Garg A, 2002 Clinical review 153: Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab 87: 4845-4856.
66. Brinkman K, Smeitink JA, Romijn JA, Reiss P, 1999 Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354: 1112-1115.
67. Behrens GM, Boerner AR, Weber K, et al, 2002 Impaired glucose phosphorylation and transport in skeletal muscle cause insulin resistance in HIV-1-infected patients with lipodystrophy. J Clin Invest 110: 1319-1327.
68. Addy CL, Gavrila A, Tsiodras S, Brodovicz K, Karchmer AW, Mantzoros CS, 2003 Hypoadiponectinemia is associated with insulin resistance, hypertriglyceridemia, and fat redistribution in human immunodeficiency virus-infected patients treated with highly active antiretroviral therapy. J Clin Endocrinol Metab 88: 627-636.
69. Coffinier C, Hudon SE, Farber, et al, 2007 HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci USA 104: 13432–13437.
70. Tran H, Robinson S, Mikhailenko I, Strickland DK, 2003 Modulation of the LDL receptor and LRP levels by HIV protease inhibitors. J Lipid Res 44: 1859-1869.
71. Bastard JP, Caron M, Vidal H, et al, 2002 Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 359: 1026-1031.
72. Gavrila A, Hsu W, Tsiodras S, et al, 2005 Improvement in highly active antiretroviral therapy-induced metabolic syndrome by treatment with pioglitazone but not with fenofibrate: a 2 x 2 factorial, randomized, double-blinded, placebo-controlled trial. Clin Infect Dis 40: 745-749.
73. Lee J, Chan J, Sourlas E, Raptopoulos V, Mantzoros C, 2006 Recombinant Methionyl Human Leptin Therapy in Replacement Doses Improves Insulin Resistance and Metabolic Profile in Patients with Lipoatrophy and Metabolic Syndrome Induced by the Highly Active Antiretroviral Therapy J Clin Endocrinol Metab 91: 2605-2611.
74. Coffinier C, Hudon S, Lee R, et al, 2008 A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl-prelamin A in cells. J Biol Chem 283: 9797-9804.
75. Norbiato G, Bevilacqua M, Vago T, Taddei A, Clerici, 1997 Glucocorticoids and the immune function in the human immunodeficiency virus infection: a study in hypercortisolemic and cortisol-resistant patients. J Clin Endocrinol Metab 82: 3260-3263.
76. Norbiato G, Bevilacqua M, Vago T, Clerici M, 1996 Glucocorticoids and interferon-alpha in the acquired immunodeficiency syndrome. J Clin Endocrinol Metab 81: 2601-2606.
77. Hadigan C, Miller K, Corcoran C, Anderson E, Basgoz N, Grinspoon S, 1999 Fasting hyperinsulinemia and changes in regional body composition in human immunodeficiency virus-infected women. J Clin Endocrinol Metab 84: 1932-1937.
78. Simpson DM, Bender AN, Farraye J, Mendelson SG, Wolfe DE, 1990 Human immunodeficiency virus wasting syndrome may represent a treatable myopathy. Neurology 40: 535-538.
79. Brown TT, Chu H, Wang Z, et al, 2007 Longitudinal increases in waist circumference are associated with HIV-serostatus, independent of antiretroviral therapy. AIDS 21: 1731-1738.
80. Kino T, Chrousos GP, 2001 Glucocorticoid and mineralocorticoid resistance/hypersensitivity syndromes. J Endocrinol 169: 437-445.
81. Wanke CA, 1999 Epidemiological and clinical aspects of the metabolic complications of HIV infection the fat redistribution syndrome. AIDS 13: 1287-1293.
82. Kino T, Chrousos GP, 2004 Human immunodeficiency virus type-1 accessory protein Vpr: a causative agent of the AIDS-related insulin resistance/lipodystrophy syndrome? Ann N Y Acad Sci 1024: 153-167.
83. Emerman M, 1996 HIV-1, Vpr and the cell cycle. Curr Biol 6: 1096-1103.
84. Kino T, Gragerov A, Valentin A, et al, 2005 Vpr protein of human immunodeficiency virus type 1 binds to 14-3-3 proteins and facilitates complex formation with Cdc25C: implications for cell cycle arrest. J Virol 79: 2780-2787.
85. Sawaya BE, Khalili K, Gordon J, Taube R, Amini S, 2000 Cooperative interaction between HIV-1 regulatory proteins Tat and Vpr modulates transcription of the viral genome. J Biol Chem 275: 35209-35214.
86. Andersen JL, Planelles V, 2005 The role of Vpr in HIV-1 pathogenesis. Curr HIV Res 3: 43-51.
87. Pavlakis GN 1996 The molecular biology of HIV-1. In: Devita VT, Hellman S, Rosenberg SA (eds) AIDS: Diagnosis, Treatment and Prevention. Lippincott Raven, Philadelphia; pp, 45-74.
88. Levy DN, Refaeli Y, Weiner DB, 1995 Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol 69: 1243-1252.
89. Sherman MP, Schubert U, Williams SA, et al, 2002 HIV-1 Vpr displays natural protein-transducing properties: implications for viral pathogenesis. Virology 302: 95-105.
90. Kino T, Gragerov A, Kopp JB, Stauber RH, Pavlakis GN, Chrousos GP, 1999 The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J Exp Med 189: 51-62.
91. Heery DM, Kalkhoven E, Hoare S, Parker MG, 1997 A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387: 733-736.
92. Kino T, Gragerov A, Slobodskaya O, Tsopanomichalou M, Chrousos GP, Pavlakis GN, 2002 Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol 76: 9724-9734.
93. Mirani M, Elenkov I, Volpi S, Hiroi N, Chrousos GP, Kino T, 2002 HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J Immunol 169: 6361-6368.
94. Shrivastav S, Kino T, Cunningham T, et al, 2008 Human Immunodeficiency Virus (HIV-1) viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor γ and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy Mol Endocrinol 22: 234-247.
95. Fawell S, Seery J, Daikh Y, et al, 1994 Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A 91: 664-668.
96. Kino T, Slobodskaya O, Pavlakis GN, Chrousos GP, 2002 Nuclear receptor coactivator p160 proteins enhance the HIV-1 long terminal repeat promoter by bridging promoter-bound factors and the Tat-P-TEFb complex. J Biol Chem 277: 2396-2405.
Address for correspondence:
Evangelia Zapanti, 8 s. Karageorga str., 166 75 Glyfada,
Greece, e-mail: liazapanti@yahoo.gr
Received 27-06-07, Revised 30-03-08, Accepted 20-04-08