Address Correspondence to:
C. Dacou-Voutetakis, First Pediatric Department, Athens University Medical School, Aghia Sophia Children’s Hospital, Goudi, 115 27 Athens, Greece, Tel.: 010.7795538, Fax: 010.7795538
Received 14-09-2001, Revised 25-11-2001, Accepted 12-12-2001
Abstract
We report a boy with pseudohypoparathyroidism (PHP), hypothyroidism and low growth hormone (GH) values with no response to growth hormone releasing hormone (GHRH). He presented at age 17 mo because of developmental delay. He had the typical features (short stature, obesity, round face, brachydactyly) of Albright’s hereditary osteodystrophy (AHO) and the biochemical profile of PHP; low serum calcium and high phosphate, raised parathormone (PTH) values and lack of response of urinary phosphate and cyclic AMP to PTH administration. The serum total thyroxine value (T4) was 37.32 nmol/L and the thyroid stimulating hormone (TSH) 29 mU/L. Peak GH values during two provocative tests (Glucagon, L-Dopa) were <2.5 μg/L and <1.7 μg/L, respectively, while following GHRH administration the maximum GH value was 0.2 μg/L. The IGFI value was 65 ng/ml and rose to 253 ng/ml after GH administration for three days. This boy had PTH and TSH receptor defect and we speculate that he also has GHRH receptor defect.
INTRODUCTION
Pseudohypoparathyroidism (PHP) comprises a heterogeneous group of genetically determined disorders. PHP is characterized by resistance to PTH at its main target organs (kidney and bone) and is classified into two types (I and II) depending on the presumed site of resistance to PTH. In PHP type I, PTH resistance is expressed as failure to elicit an appropriate increment in urinary cAMP and phosphate excretion following exogenous PTH infusion. PHP type I is subdivided into two types Ia and Ib. PHP type Ia is associated with AHO, a characteristic skeletal phenotype, which includes short stature, obesity, round face, brachydactyly, and dental hypoplasia. Patients with PHP type Ib have PTH resistance but do not have the AHO phenotype. Family members of PHP patients, who have the skeletal features of AHO but do not have the metabolic abnormalities, have been designated as having PPHP1,2. Patients with PHP may present resistance to other hormones such as gonadotrophins and thyroid stimulating hormone that stimulate cAMP formation in the target cells by interacting with receptors coupled to Gs, the stimulatory protein of adenyl cyclase3,4.
We report a 17 mo old boy with PHP type Ia, who also presented hypothyroidism and low GH values with no response to GHRH, presumably as a result of GHRH resistance.
METHODS
Serum 25(OH)VitD was measured by RIA (Nichols Inst Diagnostics, USA) normal values: 45-90 nmol/L, sensitivity: 2 ng/ml, intra and inter coefficient of variation: 8.2-10.4% and 10.8-15%, respectively. PTH was determined by IRMA (intact PTH immunoassay, Nichols Inst Diagnostics, USA), normal values: 10-65 pg/ml, sensitivity: 1pg/ml, intra and inter coefficient of variation: 1.8-3.4% and 5.6-6.1%, respectively. Urinary phosphate and urinary cAMP (RIA, Amersham Int, UK) were measured prior to and following infusion of the synthetic 1-34 aminoterminal fragment of human PTH (Parathar, teriparatide acetate, 3 U/Kg). Serum T4 was measured by FPIA (IMX, Abbott Laboratories, USA), normal values: 51-142 nmol/L, sensitivity: 1 £g/dl, intra and inter coefficient of variation: 4.5% and 2.9%, respectively. TSH was determined by MEIA (IMX, Abbott Laboratories, USA), normal values: <5 mU/L, sensitivity: 0.03μ IU/ml, intra and inter coefficient of variation: 3.8% and 5.4%. GH was measured by Chemiluminescence Immunoassay (Nichols Inst Diagnostics, USA) normal values: >10μg/L, sensitivity: 0.1 ng/ml, intra and inter coefficient of variation: 4.2-8% and 4.1-12%, respectively. Glucagon provocative test: Glucagon (100μg/kg) was given intramuscularly and blood samples for glucose and GH determinations were drawn every 30 min for 3 hours. L-DOPA provocative test: Levodopa 0.5 g/1.73m2 was administered orally at time 0 and growth hormone was measured at 0, 30, 60, 90 and 120 minutes. GHRH provocative test: GH was measured prior to and 30, 45, 60 and 120 minutes following the intravenous administration of GHRH (1mcg/kg). IGFI was measured prior to and 24, 48,and 72 hours after GH administration (0.15 IU/kg/day for 3 days). IGFI determination was carried out by Chemiluminescence Immunoassay (Nichols Inst Diagnostics, normal values males 2-5 y: 17-248 ng/ml, sensitivity: 0.1 ng/ml, intra and inter coefficient of variation: 4.4-5.2% and 5.4-7.4%. Serum LH was measured by Chemiluminescence (Chiron Diagnostics Corp, USA), normal prepubertal values <0.1 mIU/ml, sensitivity: 0.07 mIU/ml, intra-assay coefficient of variation: 5-6.3%. FSH was measured by Chemiluminescence (Bayer Corp, USA), normal prepubertal values <1.6 mIU/ml, sensitivity: 0.3 mIU/ml, intra-assay coefficient: 4.5-4.7%.
CLINICAL DATA
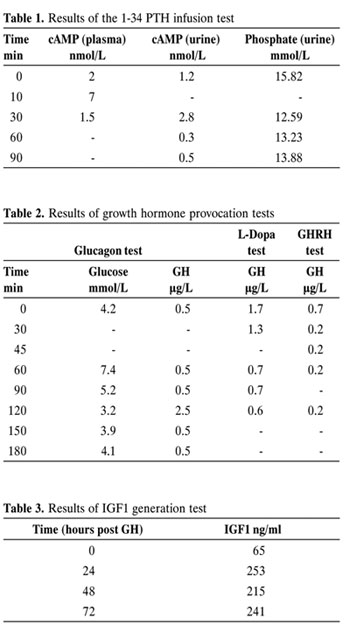
A 17 mo old male infant, the first child of unrelated Greek parents, presented because of developmental delay. He was born at term after an uncomplicated pregnancy by vaginal delivery and with a birth weight of 2850 g. On admission, physical examination revealed short stature, obesity, round face and brachydactyly. A 2/6 systolic murmur was audible at the aortic region. His weight was above the 97th percentile, whereas his height was at the 3rd and his head circumference at the 75th. The target height was 170 cm. The serum calcium values ranged from 1.54 to 1.89 mmol/L (6.2 to 7.6 mg/dl), the serum phosphate from 2.16 to 2.29 mmol/L (6.7 to 7.1 mg/dl) and the alkaline phosphatase from 0.5 to 0.6 μkat/L (30 to 37 U/L). No symptoms of hypocalcemia were reported. The serum 25(OH)VitD concentration was 57 nmol/L (23 ng/ml). The PTH values ranged from 141 to 179 pg/ml. Following PTH infusion urinary phosphate did not change and the expected increase in urinary cAMP was not observed (Table 1). The serum T4 was 37.32 nmol/L (2.9 μg/dl) and the TSH 29 mU/L (29 μU/ml). The peak GH values during glucagon and L-DOPA tests were 2.5 μg/L and 1.7 μg/L (2.5 ng/ml and 1.7 ng/ml), respectively. Following GHRH administration the maximum GH value was 0.2 μg/L (0.2 ng/ml) (Table 2). The basal IGFI level was 65 ng/ml and after GH administration rose to a maximum of 253 ng/ml (Table 3). Serum LH and FSH values were <0.1 and 1.6 IU/L (<0.1 and 1.6 mIU/ml), respectively. Blood cell count, electrolytes, urea, creatinine, glucose, magnesium, hepatic transaminases, renal ultrasound and brain computed tomography were normal. Slit lamp examination and fundoscopy were also normal. Echocardiogram revealed mild aortic valve stenosis (gradient 20 mmHg). Hand radiograph showed shortening affecting mainly the 1st, 4th and 5th metacarpals. His Griffith’s assessment at age 54 months showed a locomotor score of 38 months, personal social score of 40 months, hearing and speech of 28 months, eye – hand coordination 30 months and performance score 30-32 months. Our patient has been treated with L thyroxine, Ca and 1α˜(OH)D3 since the time of diagnosis. On this therapeutic regimen he has been normocalcemic and has normal thyroid function. At the age of 8 2/12 yrs hGH was initiated at the dose of 0.5 IU/kg/week given subcutaneously by daily injections. The annual growth velocity prior to hGH was 3.1 cm, while on hGH the annual growth velocity rose to 9.2 cm the first year and the height gradually reached the 25th percentile. The ratio of bone age to chronological age during hGH treatment was 1. Thus his height rose from the 6th to the 25th percentile on hGH therapy. The bone age (9yrs at the chronological age of 8yrs prior to hGH administration) was advanced and hence the SDS for bone age was -2.5 prior to hGH initiation. The advanced bone age is not readily explained since there was no evidence of puberty initiation (testicular volume 1.5 ml and pubic hair growth Tanner I).
DISCUSSION
Certain patients with PHP may show resistance not only to PTH but also to other hormones that act via cAMP. The clinical and hormonal profile is therefore quite heterogeneous. Thus, hypothyroidism, at least biochemical, has been reported as well as resistance to gonadotrophins leading to hypogonadism3,4. From the biochemical and clinical findings our patient must be classified as PHP type Ia. In addition to the commonly reported hypothyroidism our patient also exhibited low GH values with no response to GHRH. The GH values following GHRH administration were extremely low (peak GH value: 0.2 μg/L- 0.2 ng/ml) and the low IGFI value rose significantly after exogenous GH administration. Since GH release induced by GHRH and both spontaneous and stimulated GH secretion are reduced in patients with hypothyroidism, the GH secretory ability was studied while our patient was euthyroid with thyroxine administration. The growth pattern following GH administration constitutes additional evidence of GH insufficiency. We thus speculate that the low GH values are caused by GHRH insensitivity. GHRH acts on somatotropes by binding to a G-protein coupled receptor. It is thus surprising that GH deficiency is only rarely reported in patients with PHP type Ia5.
Most patients with PHP type Ia have a partial deficiency (50%) of Gs activity due to a reduction in its ˜-subunit (Gs˜α) mRNA and protein. The human Gsα˜ gene (GNAS1) is located at 20q13.11 and consists of 13 exons. Heterozygous mutations in GNAS1 have been identified in the majority of patients with PHP type Ia and PPHP. The dominant pattern of inheritance has been attributed to haploinsufficiency of the Gsa and the tissue-specific haploinsufficiency of Gsa explains the selective resistance to hormones6-9.
At start of hGH therapy the height for chronological age was at the 6th percentile. Nevertheless, the bone age was advanced (9 years at the chronological age of 8 years) and hence the height for bone age was below the 3rd percentile. More specifically, the standard deviation score for bone age was -2.5 prior to hGH administration. The annual growth velocity prior to hGH was 3.1 cm, while on hGH the annual growth velocity rose to 9.2 cm the first year and the height gradually reached the 25th percentile. The ratio of bone age to chronological age during hGH treatment was 1.
The advanced bone age constitutes an impressive finding in the absence of puberty. This may mask shortness, as in our case, and further compromise final height if it is not appreciated promptly.
In conclusion, the boy described here presents PTH, TSH and most likely GHRH receptor defect. Possible resistance to gonadotrophins cannot be diagnosed because the patient is in the prepubertal stage. Further observations are necessary to define the frequency and significance of GH deficiency and the role of GH replacement therapy in improving final height in patients with PHP.
REFERENCES
1. Silve C, 1995 Pseudohypoparathyroidism syndromes: the many faces of parathyroid hormone resistance. Eur J Endocrinol 133: 145-146.
2.Kruse K, 1996 Albright’s hereditary osteodystrophy. Eur J Pediatr 155: 349-350.
3. Spiegel AM, 1999 Hormone resistance caused by mutations in G proteins and G protein-coupled receptors. J Pediatr Endocrinol Metab 12(Suppl 1): 303-9.
4. Farfel Z, Bourne HR, Taroh I, 1999 The expanding spectrum of G protein diseases. N Engl J Med 340: 1012-20.
5. Scott DC, Hung W, 1995 Pseudohypoparathyroidism type Ia and growth hormone deficiency in two siblings. J Pediatr Endocrinol Metab 8: 205-207.
6. Ringel MD, Schwindiger WF, Levine MA, 1996 Clinical implications of genetic defects in G proteins. The molecular basis of McCune-Albright syndrome and Albright hereditary osteodystrophy. Medicine-Baltimore 75: 171-184.
7. Miric A, Vechio JD, Levine MA, 1993 Heterogeneous mutations in the gene encoding the alpha-subunit of the stimulatory G protein of adenyl cyclase in Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab 76: 1560-1568.
8. Davies SJ, Hughes HE, 1993 Imprinting in Albright’s hereditary osteodystrophy. J Med Genet 30: 101-103.
9. Mantovani G, Romoli R, Weber G, Brunelli V, De Menis E, et al, 2000 Mutational analysis of GNAS1 in patients with pseudohypoparathyroidism: identification of two novel mutations. J Clin Endocrinol Metab 85: 4243-8.