Corresponding and reprint request author:
Neoklis A. Georgopoulos: Department of Internal Medicine, Division of Endocrinology, University of Patras Medical School, University Hospital, Rion 26500, Patras, Greece. Tel: 0610-999582, Fax: 0610-993982, e-mail: vag.inmd@med.upatras.gr
Received 11-10-2001, Revised 16-11-2001, Accepted 15-12-2001
Abstract
Thyrotropin (TSH) is the prime regulator of thyroid cell growth and function and acts through the thyrotropin receptor (TSHR) located on the surface membrane of thyrocytes. Somatic heterozygous mutations that cause TSHR activation in the absence of TSH have been found in toxic adenomas and in hot nodules of multinodular goiters. Clinically and histologically heterogeneous nodules can share common gain-of-function mutations. Mutation prevalence varies greatly and is inversely related to iodine intake of the population. We report a Greek patient presenting with subclinical hyperthyroidism due to a fast-growing autonomous hyperplastic nodule in a long-standing multinodular goiter. Direct DNA sequencing showed that the hot nodule harbored a somatic heterozygous activating TSHR mutation: substitution of glutamine for leucine in the third transmembrane helix. This mutation (L512Q) was recently described in two solitary toxic adenomas. This report expands the spectrum of mutations shared by dissimilar hot nodules, supporting a common mechanism for nonautoimmune thyroid autonomy. The identification of the L512Q substitution demonstrates that gain-of-function TSHR mutations are encountered in Greece, although iodine deficiency has been significantly corrected over the last three decades.
INTRODUCTION
Under physiological conditions, differentiation and proliferation of thyroid cells are regulated primarily by thyrotropin (TSH)1. TSH acts by binding to the TSH receptor (TSHR) on thyrocyte membranes. TSHR belongs to the family of G protein-coupled receptors, with the typical motif of seven transmembrane segments connected by three extra- and three intracellular loops2 (Figure 1). Thyrotropin binding to TSHR activates, through Gsα protein, the cAMP cascade mainly responsible for mediating TSH effects. At higher hormone concentrations, the less important inositol phosphate pathway is also triggered via Gsα1.
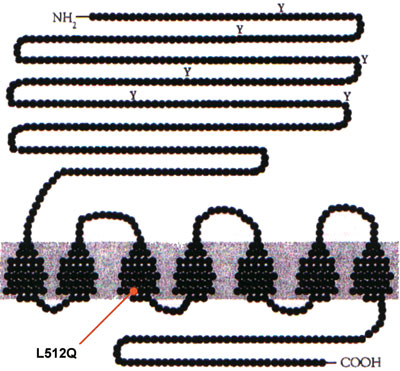
Figure 1.The thyrotropin receptor (TSHR) belongs to the family of G protein-coupled receptors, with the typical motif of seven transmembrane segments connected by three extra- and three ιntracellular loops (adapted from Russo D et al7). Leucine at position 512 lies within the third transmembrane helix of TSHR. The autonomous nodule of our patient harbors a heterozygous substitution of glutamine for lysine (L512Q).
It was predicted that somatic gain-of-function mutations affecting proteins of the TSH signalling pathway would lead to the clonal expansion of the mutated cell and the generation of a clone of autonomously growing and functioning thyrocytes. This would correspond to the clinical entity of solitary toxic adenoma3. Activating mutations were indeed identified in toxic adenomas, initially in the Gsα protein4 and later in the TSHR itself5. Mutations are somatic, heterozygous and confined to the autonomous tissue; all are located in exons 9 and 10 of the receptor gene, which encode for the intracellular, transmembrane and proximal extracellular domains2. No activating mutations were found within TSHR exons 1-8, which encode for most of the extracellular domain. Mutant receptors cause constitutive (TSH-independent) activation of the cAMP and/or inositol phosphate pathways6. The prevalence of gain-of-function TSHR mutations is highly variable among different series7. Methodological discrepancies are partly incriminated, but different iodine intake of populations also seems responsible8.
Constitutively activating TSHR mutations were subsequently found in solitary or multiple hot nodules of toxic or autonomous multinodular goiters9,10. Toxic multinodular goiter represents the final phase in the evolution of goiter over time; it develops slowly within a gland whose nodules gain autonomy3. In this clinical setting, autonomous nodules are histologically classified as either typical adenomas or hyperplastic nodules11 and gain-of-function mutations exist in both types12,13. Although not present in all hot nodules, these mutations emerge as a possible common mechanism underlying the pathogenesis of non-autoimmune thyroid autonomy6.
We report here the first activating mutation identified in a hyperplastic hot nodule of a Greek patient with autonomous multinodular goiter. By direct genomic sequencing of the TSHR exon 10 we found a single aminoacid substitution, L512Q, in the third transmembrane segment of the receptor (Figure 1). This mutation was recently identified by different methodology in two German patients with solitary toxic adenomas14.
PATIENT REPORT
A 50-year old female patient with a 10-year history of euthyroid multinodular goiter was seen in the outpatient endocrine clinic. She was on no medication. Her main complaint was a recent swelling of the right lobe of the thyroid gland. She was clinically euthyroid. Laboratory investigation showed a T3 value of 2.4 nmol/L (160ng/dL) (normal range: 1.2-3.0 nmol/L, 80-200 ng/dL), a T4 value of 92 nmol/L (7.2 μg/dL) (normal range: 58-160 nmol/L, 4.5-12.5 μg/dL) and a TSH value of 0.35 mU/L (0.35 μU/mL) (normal range: 0.4-4.0), indicating subclinical hyperthyroidism. The antiperoxidase-(TPO) and antithyroglobulin antibodies were negative. Thyroid ultrasound showed multiple nodules bilaterally within an enlarged thyroid gland (right lobe: 5.8×1.6×2.2 cm, left lobe: 4.5×1.5×1.3 cm). The dominant nodule was in the inferior pole of the right lobe, cystic in consistency and 3.5 cm in diameter. A 99m-Tc scintiscan revealed increased uptake by the dominant nodule and suppression of the remaining parenchyma. The patient was subjected to subtotal thyroidectomy. Histologically, all thyroid nodules showed a hyperplastic pattern.
Immediately after thyroidectomy, tissue samples were taken from the autonomous nodule and surrounding normal tissue. Samples were carefully excised matching the scintiscan and ultrasound patterns with the whole gland laid in its proper anatomic orientation. Tissue specimens were shock-frozen in liquid nitrogen. Peripheral blood sample was collected in EDTA and stored at -200C.
The study complied with the stipulations of the declaration of Helsinki. Informed consent was obtained from the patient before surgery.
Materials and Methods
Genomic DNA was extracted from surgical samples following the standard phenol/chlorophorm procedure and from peripheral blood leukocytes using Nucleospin Blood QuickPure kit (Macherey – Nagel, Düren, Germany). The complete exon 10 of the thyrotropin receptor gene, which encodes for the transmembrane and intracellular domains of TSHR, was amplified by polymerase chain reaction (PCR) as previously described15. Two overlapping sequences were amplified with two sets of oligonucleotide primers (MWG-Biotech AG, Ebersberg, Germany): An 875bp fragment with forward primer 5/-ACTGTCTTTGCAAGCGAGTT- 3/and reverse primer 5/-GTGTCATGGGATTGGAATGC -3/ and an 868bp fragment with forward primer 5/-ACTGTCTTTGCAAGCGAGTT -3/ and reverse primer 5/ -GTGTCATGGGATTGGAATGC- 3/. 100 ng of genomic DNA was used as template in a PTC-200 thermocycler (MJ Research Inc., Waltham, MA, USA). PCR was performed in a total volume of 50 μl, using 10 pmol of each appropriate primer, 200 μM of each dNTP, 1.5 mM MgCl, 2.5 U Gibco Brl Taq DNA polymerase (ANTI-SEL, Thessaloniki, Greece) and 10x reaction buffer containing 200mM Tris-HCl (pH=8.4) and 500 mM KCl. After an initial denaturing at 95ψC for 5 min, reactions were subjected to 30 cycles of 30 sec denaturing at 950C, annealing at 560C and extension at 720C, followed by a final 6 min extension at 720C. PCR products were purified with Nucleospin Extract kit (Macherey-Nagel, Düren, Germany). At least two different purified PCR amplicons were sequenced on both sense and anti-sense strands. Sequencing reactions were performed using ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Weiterstadt, Germany) with the PCR primers as sequencing primers and run on an ABI 373 DNA Sequencer (Applied Biosystems, Foster City, CA, USA).
Results
Direct automated sequencing of thyrotropin receptor (TSHR) exon 10 showed a T to A transversion in the heterozygous state (Figure 2) in the hyperfunctioning hyperplastic nodule. This results in the substitution of glutamine (CAG) for leucine (CTG) at codon 512 in the third transmembrane helix (Figure 1). The height of the mutant and wild-type base signal peaks was almost identical (Figure 2). This indicates that the sample consisted purely of tissue heterozygous for the L512Q mutation. The substitution was absent from surrounding non-autonomous tissue and peripheral blood leukocytes (Figure 2), proving that the mutation was somatic. This is a gain-of-function mutation recently described in two German patients harboring solitary toxic adenomas14. When transfected in COS7 cells14, the mutant receptor reached 87% of the wild-type TSHR cell surface expression level. It promoted a basal cAMP accumulation five times higher than wild-type receptor and a TSH-stimulated response slightly less than wild-type.
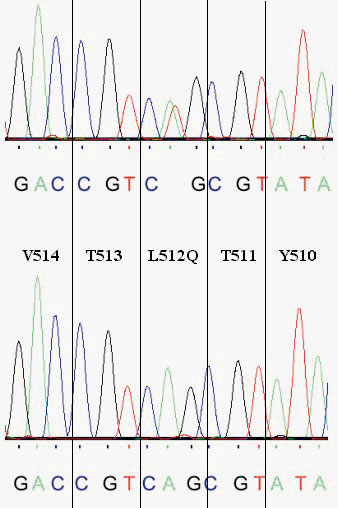
Figure 2.Chromatograms of the anti-sense strands of TSHR exon 10 PCR amplicons. Automated genomic sequencing of DNA extracted from the autonomous nodule revealed a heterozygous T–>A transversion (top chromatogram). This causes the substitution of glutamine (cag) for leucine (ctg) at aminoacid 512 in the third transmembrane helix of the thyrotropin receptor. The mutant and wild-type base signals show identical peaks, indicating absence of contamination with wild-type sequence from normal tissue. Sequencing of DNA extracted from nonautonomous tissue or peripheral blood leukocytes demonstrates the wild-type sequence only (bottom chromatogram), proving that the mutation is of somatic nature.
Discussion
Somatic heterozygous gain-of-function mutations of the thryrotropin receptor (TSHR) and Gsα protein were initially described in solitary toxic adenomas of the thyroid gland4,5. These mutations cause TSH-independent (constitutive) activation of the cAMP and/or inositol phosphate signalling pathways6, which control thyrocyte growth and function1. Activating mutations were subsequently identified in hot nodules of toxic or autonomous multinodular goiters9,10. The term “toxic multinodular goiter” (TMG) encompasses a wide spectrum of different clinical entities, ranging from a single hyperfunctioning nodule within an enlarged thyroid gland having additional nonfunctioning nodules, to multiple hyperfunctioning areas scattered throughout the gland, barely distinguishable from nonfunctioning nodules and extranodular parenchyma3,16. Moreover, hyperfunctioning nodules may present histologically as typical adenomas (encapsulated homogeneous follicular structures) or as hyperplastic nodules (micro- macrofollicular aggregates devoid of capsule)11. Constitutively activating TSHR and Gsα mutations have been found in autonomous tissues of all these clinical and pathologic variants12,13. The L512Q mutation was recently reported in two German patients with toxic uninodular goiter, whose hot nodules showed the typical histological presentation of adenoma14. We have identified the same mutation in a hyperplastic hot nodule of a patient with autonomous multinodular goiter. This expands the spectrum of mutations shared by autonomous nodules of different clinical and histological backgrounds. Thus, it reinforces the concept of a common mechanism underlying the pathogenesis of non-autoimmune autonomy6.
Functional studies of the L512Q TSHR revealed a constitutive activation of the cAMP cascade five-fold over the wild-type receptor14. Although the signalling through the PIP2 phospholipase-C cascade was not studied, in comparison to other activating TSHR mutations17, this represents a relatively strong constitutive activity. Our patient presented with subclinical hyperthyroidism due to a recently growing nodule in a long-standing multinodular goiter. This is in accordance with L512Q driven stimulation of thyrocyte growth and function. However, the clinical characteristics of patients with autonomous nodules harboring activating mutations are not necessarily associated with receptor constitutive activity6. Identical somatic or germline mutations can present with highly variable clinical pictures6. This implies that other factors are involved in the clinical expression of an activating event in the TSHR signalling pathway8, including alterations of the G-protein signalling or overexpression of the TSHR itself due to defects in receptor desensitization and internalization18.
Interestingly, in the two adenomas previously reported to harbour the L512Q substitution14, initial direct sequencing of the nodules’ genomic DNA failed to identify the mutation. The mutation was subsequently found by direct sequencing of heteroduplex bands formed in a denaturing gradient gel electrophoresis (DGGE). This demonstrated the ability of DGGE to recognize the presence of heterozygous mutations against a higher background of wild type sequence19. This is important when heterozygous somatic mutations are sought, because tissue samples may contain fibroblasts, blood leukocytes, or surrounding normal tissue. Such contamination decreases the percentage of the mutant allele in the sample below the ideal 50%, and possibly below the detection limit of direct automated sequencing. This has been advocated as one of the reasons for the highly variable prevalence of TSHR and Gsα mutations in different series7. The identical height of the mutant and normal base peaks (Figure 2), indicates the absence of contamination in our sample. This emphasizes the importance of meticulous sample selection in conjunction with a sensitive detection method.
Codon 512 in the third transmembrane helix (TM3) of TSHR emerges as a receptor site frequently mutated in autonomous nodules. L512Q has been found in three cases, including this report14. Another activating substitution at the same codon, L512R, has been reported twice14,20. TM3 encompasses the TSHR highly conserved aminoacids 497-517. Naturally occurring activating mutations in TM3 also affect aminoacids 505 and 509. In vitro mutagenesis of residues 505-513 demonstrated that mutations at positions other than 505, 509 and 512 do not induce constitutive receptor activation20. These three aminoacids are presumably located on the surface of a simple ˜-helix, facing in the same direction. They may be involved in formation of non-covalent bonds with another helix that participate in maintaining the receptor in a constrained inactive conformation20. In contrast to initial reports identifying mutations within or near the third intracellular loop5, it is now evident that activating mutations are scattered throughout the carboxyl half of the receptor6. This emphasizes the need for screening completely the corresponding exon 10 of the TSHR gene. The identification of frequently affected residues provides insight into the TSHR structure-function relationship and the mechanisms of receptor activation.
Gain-of-function mutations have been reported with varying frequencies in different series of solitary toxic adenomas, ranging from 0% to 82%7. Methodological reasons may partly account for such extreme discrepancies. Interestingly, the prevalence of activating mutations is inversely related to iodine intake of the population8, as is the prevalence of TMG3. Here we report the first TSHR mutation found in a Greek patient with sub-toxic multinodular goiter. Iodine deficiency was well documented in post-war Greece21, but has been corrected over the last 30 years due to increased use of iodized salt and improved socioeconomic conditions22,23. Although iodine deficiency still persists in small mountainous regions23, particularly in areas of Southwestern Greece24, the prevalence of goiter has accordingly decreased21,24. TMG represents the final stage in the evolution of goiter over time and its prevalence increases with age3,16. Therefore elderly patients have probably been affected by iodine deficiency at a younger age. Thus, the prevalence of gain-of-function mutations in autonomous nodules in Greece is likely to be intermediate between the extremes reported in the literature, owing to amelioration of iodine deficiency over the last three decades.
Acknowledgment
We thank Dr Karavias, Assoc. Professor, Department of Surgery, Patras University Hospital, for provision of tissue samples.
This study was supported by a “Karatheodoris” grant.
REFERENCES
1. Dumont JE, Lamy F, Roger P and Maenhaut C, 1992 Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev 72: 667-697.
2. Nagayama Y and Rapoport B, 1992 The thyrotropin receptor 25 years after its discovery: new insight after its molecular cloning. Mol Endocrinol 6: 145-156.
3. Siegel RD and Lee SL, 1998 Toxic nodular goiter. Toxic adenoma and toxic multinodular goiter. Endocrinol Metabolism Clin North Am 27: 151-168.
4. Lyons J, Landis CA, Harsh G, et al, 1990 Two G protein oncogenes in human endocrine tumors. Science 249: 655-659.
5. Parma J, Duprez L, Van Sande J, et al, 1993 Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365: 649-651.
6. Vassart G, 1997 New pathophysiological mechanisms for hyperthyroidism. Horm Res 48(S4): 47-50.
7. Russo D, Arturi F, Chiefari E, Filetti S, 1999 Thyrotropin receptor: a role for thyroid tumourigenesis? Forum (Genova) 9: 166-175.
8. Derwahl M, 1996 TSH receptor and Gs-alpha gene mutations in the pathogenesis of toxic thyroid adenomas–a note of caution. J Clin Endocrinol Metab 81: 2783-2785.
9. Holzapfel HP, Fuhrer D, Wonerow P, et al, 1997 Identification of constitutively activating somatic thyrotropin receptor mutations in a subset of toxic multinodular goiters. J Clin Endocrinol Metab 82: 4229-4233.
10. Tonacchera M, Chiovato L, Pinchera A, et al, 1998 Hyperfunctioning thyroid nodules in toxic multinodular goiter share activating thyrotropin receptor mutations with solitary toxic adenoma. J Clin Endocrinol Metab 83: 492-498.
11. Hedinger C, Williams ED and Sobin LH, 1988 Histological Typing of thyroid tumors. In: WHO. International histological classification of tumors. Berlin: Springer: 5-6.
12. Tonacchera M, Vitti P, Agretti P, et al, 1998 Activating thyrotropin receptor mutations in histologically heterogeneous hyperfunctioning nodules of multinodular goiter. Thyroid 8: 559-564.
13. Tonacchera M, Agretti P, Chiovato L, et al, 2000 Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 85: 2270-2274.
14. Trulzsch B, Krohn K, Wonerow P, et al, 2000 Detection of thyroid-stimulating hormone receptor and Gs alpha mutations: in 75 toxic thyroid nodules by denaturing gradient gel electrophoresis. J Mol Med 78: 684-691
15. de Roux N, Misrahi M, Chatelain N, et al, 1996 Microsatellites and PCR primers for genetic studies and genomic dequencing of the human TSH receptor gene. Mol Cell Endocrinol 117: 253-256.
16. Hay ID, Morris JC, 1996 Toxic adenoma and toxic multinodular goiter. In: Braverman LE, Utiger RD (eds) Werner and Ingbar’s The Thyroid. Philadelphia: Lippincott-Raven: 566-572.
17. Van Sande J, Parma J, Tonacchera M, et al, 1995 Somatic and germline mutations of the TSH receptor gene in thyroid diseases. J Clin Endocrinol Metab 9: 2577-2585.
18. Paschke R, Ludgate M, 1997 The thyrotropin receptor in thyroid diseases. N Engl J Med 337:1675-1681.
19. Trulzsch B, Krohn K, Wonerow P and Paschke R, 1999 DGGE is more sensitive for the detection of somatic point mutations than direct sequencing. Biotechniques 27: 266-268.
20. Kosugi S, Hai N, Okamoto H, Sugawa H, Mori T, 2000 A novel activating mutation in the thyotropin receptor gene in an autonomously functioning thyroid nodule developed by a Japanese patient. Eur J Endocrinol 143: 471-477.
21. Koutras DA, Piperingos G, Mantzos J, et al, 1993 Iodine nutrition and iodine deficiency disorders in Greece: signs of improvement. In: Delange F et al, eds. Iodine deficiency in Europe. New York: Plennum Press: 421-426.
22. Doufas AG, Mastorakos G, Chatziioannou S, et al, 1999 The predominant form of non-toxic goiter in Greece is now autoimmune thyroiditis. Eur J Endocrinol 140: 505-511.
23. Tsatsoulis A, Johnson EO, Andricula M, et al, 1999 Thyroid autoimmunity is associated with higher urinary iodine concentrations in an iodine-deficient area of Northwestern Greece. Thyroid 9: 279-283.
24. Markou K, Michalaki M, Makri M, et al, 1996 Iodine intake and thyroid function in villagers and city dwellers in Southwestern Greece (SWG). Thyroid 6(S1): 79.