HORMONES 2004, 3(4):259-265
DOI: 10.14310/horm.2002.——
Address correspondence and requests for reprints to:
Dr Gregory Kaltsas, “George Gennimatas” General Hospital, Leof. Mesogion 154, Holargos 11527, Athens, Greece, Tel: +30 210 7796043, Fax: +30 210 7779146, e-mail: gkaltsas@acci.gr
Received 16-09-04, Revised 24-09-04, Accepted 27-09-04
Abstract
Growth without growth hormone (GH) has occasionally been described in patients with organic pituitary pathology, and even more rarely in patients with idiopathic pituitary hormone deficiency. The mechanism of growth without GH remains a mystery. We describe a 17-year old male who grew 38.5 cm in height over a 7-year period, despite the fact that he had established panhypopituitarism. The hypopituitarism was initially attributed to a presumptive hypothalamic hamartoma which was not, however, confirmed on subsequent and prolonged follow-up. Regular endocrine evaluation confirmed persistent anterior and posterior pituitary hormonal deficiencies with severe concomitant hyperinsulinemia as shown by an exaggerated insulin response to a standard oral glucose tolerance test. In our patient, a postulated mechanism could be the severehyperinsulinemia, acting either through the insulin and/or IGF receptors and thus potentiating the mitogenic effect. This case illustrates that final height attainment within or above target height may occur in patients with idiopathic pituitary hormonal deficiency despite persistent, severe GH insufficiency.
Key words: Growth, Insulin, Obesity, Pituitary
INTRODUCTION
Normal linear growth in children along the percentile of their genetic potential is a highly complex process, regulated by the interaction of genetic, nutritional, environmental and hormonal factors1. Among the various hormonal factors implicated, growth hormone (GH) plays a predominant role and influences the growth pattern throughout the growth period2,3. Lack of GH is characterized mainly by growth retardation, short stature and by reduced growth spurt during puberty4. The vast majority of these abnormalities are restored with administration of GH1. Growth without GH is a rare but well-described phenomenon that was initially reported either following surgical interventions for suprasellar tumors, mainly craniopharyngiomas, or even without an organic cause1,5. Obesity and hyperphagia, as well as the concomitant hyperinsulinemia or a synergistic action of the post-surgery hyperprolactinemia were initially implicated for the growth spurt in children with apparent GH deficiency (GHD) following surgery of the hypothalamo-pituitary system (HPS)6,7. Later, the same phenomenon was observed in cases with empty sella syndrome8 and septo-optic dysplasia9. In such cases, it has been hypothesized that potent growth factors10-12 and GH variants13 likely mimic the biologic activity of GH thus accounting for this phenomenon. Moreover, during fetal life, growth without GH is a normal condition possibly occurring through an interplay between fetal plasma insulin and IGF-I levels14.
We here in report a distinct growth pattern in a patientwith idiopathic panhypopituitarism who has been followed for a 7-year period.The various associated clinical conditions and the proposed mechanisms for the paradox of growth in the absence of GH are also reviewed.
PATIENT REPORT
A 17-year old male was admitted for investigation of delayed puberty with a presumptive diagnosis of hypothalamic hamartoma. Hamartoma was originally suspected at the age of 10 years when the patient was examined following the acute onset of polyuria and polydipsia. The diagnosis of central diabetes insipidus (DI) was then made which subsequently responded to treatment with intranasal desmopressin (10-12 µg daily). Clinical examination at that time revealed a rather overweight child (body weight of 52 kg, 93rd percentile, Figure 1a) with mainly truncal obesity. The height was 147cm (85th percentile, Figure 1b). Radiography of the left hand revealed a bone age of 11.5years; the pubertal stage was considered “appropriate for the chronological age of the patient”. Further investigation revealed lower than the age-adjusted prolactin (PRL), insulin growth factor (IGF-I), and insulin growth factor binding protein 3 (IGFBP-3) levels. Gonadotropins, thyroid hormone, thyrotropin and basal cortisol as well as 17-hydroxyprogesterone and testosterone levels were within those predicted for his age limits (actual values are not available as all investigations were carried out in the Virchow – Klinikum Institution of Berlin, Germany). No further basal or dynamic investigations of the pituitary function were undertaken at that time. Magnetic resonance imaging (MRI) of the pituitary reportedly demonstrated a 10x8x6 mm hypothalamic mass that presented the radiological features of a hypothalamic hamartoma (original scans not available). The patient was discharged receiving intranasal desmopressin for the treatment of DI. When the patient was re-assessed at the same institution a year later, the clinical examination revealed stage 1 genital development (G1), stage 2 pubic hair (P2), a testicular volume of 2-3 ml, and a height of 152 cm. Endocrine investigation was consistent with central hypothyroidism [free thyroxine (FT4) 2.5pmol/L (normal range 9-26 pmol/L) and TSH 3.65 mU/L (normal range 0.5-5,0 mUL)] and the patient was given levothyroxine 100µg/day. Further endocrine assessment revealed no GH response following the intravenous administration of growth hormone releasing hormone (GHRH) consistent with the diagnosis of GH deficiency, while the levels of both gonadotropins [follicle stimulating hormone (FSH) and luteinizing hormone (LH)] hormones were undetectable. No further dynamic endocrine tests were performed. A subsequent pituitary MRI revealed the previously noted hypothalamic lesion which had not changed in size. At the age of 13 years, the patient was re-evaluated at the same department. Clinical examination revealed a body weight of 76.5 kg and a height of 166 cm [body mass index (kg/m2) BMI=27.8]; pubertal stage was still G1 and P2. Endocrinological investigation revealed IGF-I levels of 39 µg/L (normal age adjusted levels 55-216 µg/L) and IGFBP-3 levels of 2.18 mg/L (normal range 3-5.6 mg/L). The patient also underwent basal and dynamic endocrine investigation which revealed panhypopituitarism [inadequate GH and cortisol response to an insulin tolerance test (ITT), no TSH response to TRH, although PRL levels rose from 3.5 ng/mL to 7.5 ng/mL; LH and FSH levels rose to 2.3mU/ml, and 2.5mU/ml, respectively, following stimulation with GnRH]. A pituitary MRI was repeated, which, in contrast to the previously noted hypothalamic mass, revealed no abnormal findings. As this investigation was also performed at the same institution in Germany the scans are not available. Following the diagnosis of hypopituitarism, the patient was initiated on treatment with hydrocortisone 20 mg a.m. and 10 mg p.m., while the dose of levothyroxine was increased to 150µg/day and he continued to receive intranasal desmopressin (10-12 µg daily). No more data are available regarding either the patient’s possible adrenarche or the investigation of other metabolic actions of GH.
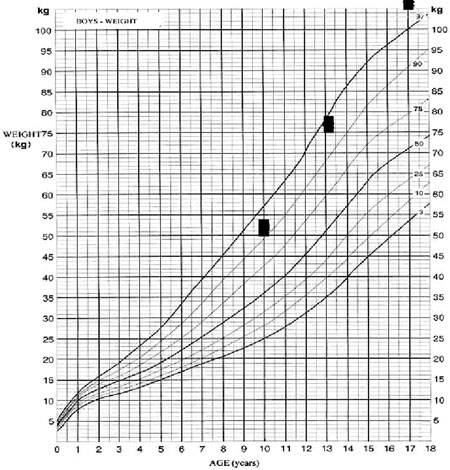
Figure 1a. Body weight (kg) of the patient from 10 to 17 yrs.
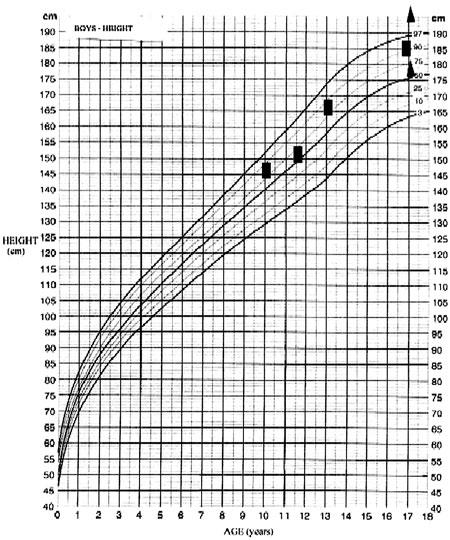
Figure 1b. Height of the patient from 10 to 17 yrs. s denotes the range of target height.
The patient was lost to follow-up and no validated data exist concerning the interim growth rate. Four years later, at the age of 17 years, he was admitted for the first time to our department due to lack of pubertal development and for re-evaluation of the previously noted hypothalamic lesion. On admission, he had a blood pressure of 100/60 mmHg. His height was 185.5 cm (predicted height:186,5 ±8 cm) and is expected to increase further since his bones are still open. He was noted to be overweight [body weight of 108kg, BMI of 32 kg/m2 (normal range 25-30)], with a waist to hip ratio (W/H) of 1.25 (normal range <1). At that time the patient was receivingintranasal desmopressin 10-12 µg daily, hydrocortisone 20 mg a.m. and 10 mg p.m., and levothyroxine 150µg/day. Evaluation of his pubertal stage revealed a genital stage of 1, pubic hair of 1-2 and a testicular volume of 2 ml bilaterally; there was no evidence of acanthosis nigricans. Laboratory evaluation showed a mild normocytic anaemia with a raised ESR and increased cholesterol (7.52 mmol/L, normal limits: <5.17 mmol/L) and urate (4.6 mmol/L, normal limits: 2.1-4.3 mmol/L) levels (Table 1 ). Basal endocrine investigation revealed adequately replaced thyroid function with the presence of positive autoantibodies, hypogonadotrophic hypogonadism, low basal cortisol and IGF-I levels and very low sex hormone binding globulin (SHBG) levels (Table 2). The maximum GH response to insulin induced hypoglycemia was 0.1µg/L, consistent with the diagnosis of GHD; in addition, a standard 75 g oral glucose tolerance test (OGTT) revealed normal glucose tolerance with concomitant severe hyperinsulinemia (Table 2). There was no evidence of abnormal IGF-II or pro-IGF II molecules (analysis was performed by courtesy of St Bartholomew’s Hospital). Roentgenograms revealed a bone age of 14 years, whereas bone density showed osteopenia (t-score of -1.54). A pituitary MRI revealed a partially empty sella with pituitary compression and stalk atrophy but no evidence of the previously noted hypothalamic pathology (Figure 2). No other evidence of GH deficiency was present, whereas an AGHDA (Adult Growth Hormone Deficiency Assessment) score was normal and did not reveal any alterations of the quality of life that could be attributed to GHD.
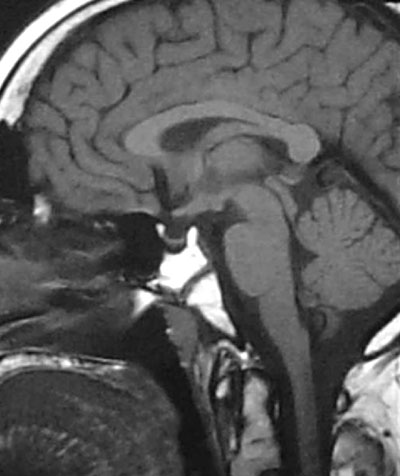
Figure 2. MRI – T1W sagittal image. Partially empty sella with pituitary compression and stalk atrophy
The diagnosis of panhypopituitarism was reconfirmed and the patient, in addition to his other pituitary hormonal deficiency replacement therapy, was also started on androgen replacement therapy with testosterone enanthate (testosterone initially 125 mg monthly and thereafter 250 mg every 20 days). Á calorie-restricted diet was also initiated while the decision for possible GH replacement treatment to ameliorate the dyslipidemic profile and improve the patient’s body composition (especially the abdominal accumulation of adipose tissue) was postponed for the follow-up period. Further growth in height is expected considering the fact that his epiphyses were still open during his last visit.
DISCUSSION
Traditionally, GH has been considered as a prerequisite for normal post-natal growth and for the achievement of a predicted final height. However, cases of normal growth without GH have previously been reported, particularly after surgery for craniopharyngioma5-7,15-18. The common finding in the majority of these cases was post-operative weight gain, which in some cases reached the point of morbid obesity. Obesity-induced hyperinsulinemia and/or the co-existence of hyperprolactinemia have been considered as possible pathogenic mechanisms6,7,15,18. A few cases of sustained growth in patients with idiopathic combined pituitary deficiency have also been described1. Similarly, our patient, although exhibiting the clinical (obesity, abnormal body composition with accumulation of abdominal fat) and endocrinological (subnormal basal IGF-I and subnormal GH levels following insulin induced hypoglycemia) features of GH deficiency, achieved substantial growth and even exceeded the expected target height. Another unique finding of this case is the maintenance and even increase in growth rate that was sustained throughout the growth period. This is in contrast to patients who display growth without GH following brain surgery, in whom the GH-independent growth is not sustained throughout the growth period1. In some of these patients GH had to be administered to achieve a normal final height1,19.
Sustained and/or accelerated linear growth has also been reported not only after surgical intervention for craniopharyngiomas or other suprasellar lesions1 but also in a number of other diseases and hypothalamic disorders5,20 and in association with some intracranial anomalies such as septo-optic dysplasia1. As most of these patients were obese, the concomitant hyperinsulinemia has again been implicated as a probable pathogenic mechanism6-13,21,although the presence of hyperprolactinemia and/or other bioavailable GH/IGF-I variants have alternatively been suggested5,12. However, it is unlikely that hyperprolactinemia could significantly affect the growth pattern5, even in cases following neurosurgical intervention where the prolactin levels remain mildly elevated22,23. Also postulated is the presence of an unidentified growth factor that could influence chondrocyte proliferation through mechanisms other than GH1. In favour of this view is the finding of a recent study in which dissociation between the growth effects of GH or another factor maintaining growth and the GH-dependent metabolic effects was documented21. This finding also raises the issue of administering recombinant GH replacement in patients exhibiting growth without GH in order to correct the possible metabolic sequellae related to the lack of GH20,21 in these children.
Recently, elevated leptin levels were found in cases with normal growth rate and GH deficiency1,24; it was thus suggested that leptin might contribute to linear growth independent of GH by directly influencing the growth plate1 since osteoblast cell proliferation was not confirmed in response to leptin24. However, the role of leptin as a regulatory signal of GH secretion remains to be identified24,especially in obese, GH deficient subjectsfollowingcraniopharyngioma removal25since the correlationof leptin with obesity and body mass index is an otherwise adequately described phenomenon24. Additionally, growth without GH was also observed in a patient with deficiencies of GH, LH and FSH, and a hypoplastic pituitary due to a mutation in the PROP1 gene26.
In our patient, the obvious hypothalamic damage did not result from a neurosurgical procedure but could be related to the initially reported hypothalamic hamartoma. Although the latest pituitary MRI revealed a partially empty sella with pituitary compression and stalk atrophy without any evidence of a hamartoma, we must assume that either there was a structural abnormality of the HPS resulting in anterior and posterior pituitary deficiency or that this could be related to a developmental defect. However, the possibility of hypothalamic damage is plausible as the patient developed obesity, probably because of increased appetite and hyperphagia as well as severe subsequent insulin resistance and hyperinsulinemia (Table 2 ). Although the presence of other, unmeasured, growth promoting factors and/or other still unrecognized factors cannot be excluded in this case, the most contingent postulated mechanism for growth without GH is the severehyperinsulinemia acting either through the insulin receptor potentiating its mitogenic effect and/ or directly affecting increasing IGF-I bioavailability.
In summary, this case illustrates that growth without GH can occur even in patients not undergoing a surgical intervention in the hypothalamo-pituitary area. Normal levels of circulating GH and/or IGF-I are not mandatory to maintain normal bone elongation. Although unknown growth factors with sufficient compensatory ability may be involved, the coexistent severe insulin resistance and compensatory hyperinsulinemia should always be investigated as insulin may be a direct stimulatory factor exerting such a mitogenic activity.
REFERENCES
1. Lazar L, Dan S, Phillip M, 2003 Growth without growth hormone: growth pattern and final height of five patients with idiopathic combined pituitary hormone deficiency. Clin Endocrinol 59: 82-8.
2. Underwood LE, Tanner JM, 1992 Normal and aberrant growth. In: Wilson JD, Foster DW (eds) Williams Textbook of Endocrinology8th ed, W.B. Saunders, Philadelphia, pp. 1079_1138.
3. Rosenfeld RG, Cohen P, 2002 Disorders of growth hormone/insulin-like growth factor secretion and action. In:. Sperling MA (ed.)Pediatric Endocrinology, 2nd edn., W.B. Saunders, Philadelphia, pp. 211_288.
4. Goodman HG, Grumbach MM, Kaplan SL, 1968 Growth and growth hormone. II. Comparison of isolated growth hormone deficiency and multiple pituitary hormones deficiency in 35 patients with idiopathic hypopituitary dwarfism. N Engl J Med 278: 57_68.
5. Geffner ME, 1996 The growth without growth hormone syndrome Haematol Oncol Clin North Am 25: 649-663.
6.Holmes LB, Frantz AG, Rabkin MT, et al, 1968 Normal growth with subnormal growth hormone levels. N Engl J Med 279: 559.
7.Kenny FM, Iturzaeta NF, Mintz D, et al, 1968 Iatrogenic hypopituitarism in craniopharyngioma: unexplained catch-up growth in three children.J Pediatr 72: 766-75.
8. Karaviti LP, 1994 Normal growth without growth hormone in a case with hypoglycemia and empty sella syndrome. Pediatr Res 33: 6276: 602.
9. Petrykowski WV, Blum WF, Ranke MB, et al, 1993 16 years of normal growth without growth hormone(GH), without IGF-I and IGFBP-3, and without obesity. Pediatr Res 33: 62.
10.Geffner ME, Lippe BM, Bersch N, et al, 1986 Growth without growth hormone: evidence for a potent circulating human growth factor. Lancet 1: 343-7.
11. Ohyama Y, Shibayama K, Yokota Y, et al. 1991 Mechanisms of growth in “growth without growth hormone ” Acta Pediatr Scand 377 :159.
12.Bistritzer T, Chalew SA, Lovchik JC, Kowarski AA, 1988 Growth without growth hormone: the “invisible” GH syndrome. Lancet 1: 321-3.
13.Finkelstein JW, Kream J, Ludan A, Hellman L, 1972 Sulfation factor (somatomedin): an explanation for continued growth in the absence of immunoassayable growth hormone in patients with hypothalamic tumors. J Clin Endocrinol Metab 35: 13-7.
14. Hill DJ, Milner RDG, 1985 Insulin as a growth factor. Pediatr Res 19: 879.
15. Bucher H, Zapf J, Torresani T, Prader A, Froesch ER, Illig R, 1983 Insulin-like growth factors I and II, prolactin, and insulin in 19 growth hormone-deficient children with excessive, normal, or decreased longitudinal growth after operation for craniopharyngioma. N Engl J Med 309:1142-6.
16. Matson DD, 1962 Craniopharyngioma. Clin Neurosurg 10: 116.
17. Frasier SD, Smith FG Jr, 1968 Return of normal growth following removal of a craniopharyngioma. Am J Dis Child 116: 311-4.
18. Silverman LA, Grumbach MM, Conte FA, 1993 Growh and acromegaloid features in a hypophysectomised (HY) male: The role of prandial hyperinsulinism (HI) Pediatr Res 33: 65.
19. Tiulpakov AN, Mazerkina NA, Brook CGD, Hindmarsh PC, Peterkova VA, Gorelyshev SK, 1998 Growth in children with craniopharyngioma following surgery. Clin Endocrinol 49: 733-738.
20. Adachi M, Tachibana K, Kigasawa H, Suwa S, 1999 “Growth without Growth Hormone” in a Young Female with Remitted Langerhans Cell Histiocytosis: A Case Report Clin Pediatr Endocrinol 8: 17-22.
21. Pavlou M, Tsatsoulis A, Efstathiadou Z, Bitsis S, Papadopoulou ZL, 2001 A study of the growth-promoting and metabolic effects of growth hormone (GH) in a patient with the “growth without GH” syndrome. Growth Horm IGF Res 11: 225-30.
22. Csizmadi I, Brazeau P, Serri O, 1989 Effect of dietary restriction and repeated growth hormone-releasing factor injections on growth hormone response to growth hormone-releasing factor in obese subjects. Metabolism 38: 1016-21.
23. Stahnke N, Grubel G, Lagenstein I, Willig RP, 1984 Long-term follow-up of children with craniopharyngioma. Eur J Pediatr 142: 179-85.
24. Hathout EH, Baylink DJ, Mohan S, 1999 Normal growth despite GH, IGF-I and IGF-II deficiency. Growth Horm IGF Res 9: 272-7.
25. Phillip M, Moran O, Lazar L, 2002 Growth without growth hormone. J Pediatr Endocrinol Metab 5: 1267-72.
26. Arroyo A, Pernasetti F, Vasilyev VV, Amato P, Yen SS, Mellon PL, 2002 A unique case of combined pituitary hormone deficiency caused by a PROP1 gene mutation (R120C) associated with normal height and absent puberty. Clin Endocrinol 57: 283-91.