Abstract
Glucocorticoids (GC) affect virtually all organ systems, acting mainly via the glucocorticoid receptor (GR). The immune system is the best characterized tissue for assessing GC sensitivity. It is well established that the immune system GC sensitivity varies widely between normal subjects. However, it remains unclear whether measurements of the immune system GC-sensitivity reflect the GC-sensitivity in other GC target tissues of the same individual. Thus, in the present study we compared the GC sensitivity of the immune system, assessed by determining the dexamethasone inhibition of LPS-induced TNF-a production in peripheral leukocytes, with the feedback sensitivity of the HPA axis, assessed by a very low dose dexamethasone (0,25mg) suppression test, in sixteen healthy volunteers. We observed a wide variation in the magnitude of the responses in the two GC targets. However, and in agreement with a number of previous reports, in a given subject the GC sensitivity of the immune system did not correlate with that of HPA axis inhibition, indicating a tissue specificity of GC sensitivity in the same individual. In summary, the bulk of current evidence suggests that GC sensitivity is tissue specific for a given individual. Additional studies are warranted to elucidate the exact mechanism(s) involved in the differential GC tissue responsiveness.
INTRODUCTION
Glucocorticoids (GC) act on virtually all organ systems, regulating a large number of metabolic, cardiovascular and immune functions. Most of their effects are mediated by binding to the glucocorticoid receptor (GR). The magnitude of the response to GCs depends on several parameters such as the concentration of the free hormone, its relative potency for binding to GR and the ability of the target cell to receive and transduce the hormone signal1. Alterations in any of these factors may result in individual differences in glucocorticoid sensitivity. Current evidence suggests that there is a wide variation in GC sensitivity amongst humans2-7. Thus, from clinical observations it has been known for many years that patients requiring GC treatment for various disorders do not exhibit the same sensitivity both with regard to efficacy and to the prevalence and severity of side effects8-10. In recent studies, laboratory assessment of GC sensitivity in various target tissues confirmed the wide variation of GC sensitivity among healthy volunteers4,6,7,11. The best characterized tissue for assessing GC sensitivity is the immune system where GCs affect cell proliferation and inhibit the release of the proinflammatory cytokines IL-6 and TNF-a11-13. Several studies have shown that the intrinsic sensitivity of an individual’s immune system to GC-mediated suppression is a major factor in determining its response to steroid therapy for inflammatory conditions8,13,14.
Thus, measurements of the immune system GC-sensitivity may become a useful tool in predicting the efficacy of steroid therapy on the disease outcome. However, it is currently unclear whether measurements of the immune system GC-sensitivity may also predict the extent of GC effects in other susceptible target tissues of the same individual. In fact, studies investigating whether the GC sensitivity of the immune system reflects the GC sensitivity of other organs in the same subject led to controversial results.
The aim of the present study was to assess whether the immune GC sensitivity assessed in peripheral blood cells is a characteristic of all target tissues in an organism or is tissue-specific, as a number of previous studies have implicated. The hypothalamo-pituitary adrenal (HPA) axis is a well-known target of GCs, in which GCs exert a strong feedback inhibition. Various types of dexamethasone suppression tests are routinely used to differentiate between normal and abnormal function of the HPA axis. For this purpose doses of dexamethasone that produce maximum suppression of the HPA axis are commonly used. By using lower doses of dexamethasone, however, a wide variation of post-dexamethasone cortisol levels are observed in normal volunteers. Based on recent suggestions the degree of suppression of cortisol levels by very low doses of dexamethasone can be used as an index of GC feedback sensitivity (4). In this study, therefore, we compared the GC sensitivity of the immune system, assessed in peripheral blood cells, as previously described, with the feedback sensitivity of the HPA axis, assessed by a very low dose dexamethasone suppression test, in healthy adults.
SUBJECTS AND METHODS
Subjects
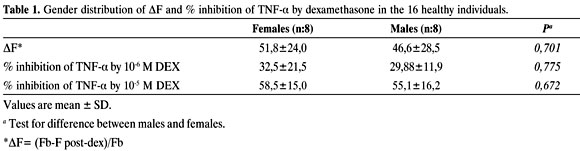
Sixteen healthy volunteers (8 men, mean age 29.63±4.81 years, BMI 25.03±4.07 and 8 women, mean age 38.88±13.4 years, BMI 23.79±4.51), medication free, participated in this study. Subjects reported to our laboratory on two consecutive days. On the first day, blood samples were collected after an eight-hour fast, between 08.30 and 09.00, for the assessment of sensitivity to GCs and for the measurement of basal cortisol levels. In addition, screening laboratory chemistry and hematology tests were performed and the basal levels of TNF-a, C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were measured to exclude a possible inflammatory process that would interfere with our study. Subjects were instructed to take 0.25mg dexamethasone at 23.00 and next day blood was drawn between 8.00 and 8.30 to determine post-dexamethasone cortisol levels.
Sample collection
For the assessment of sensitivity to GCs and for the measurement of basal TNF-a levels, venous blood was collected in endotoxin-free heparinized tubes (no-6541, Becton and Dickinson, Rutherford, NJ, USA). All samples were processed within 45 min. Serum was used for cortisol determinations.
TNF-a stimulation assay
The assay is based on the suppression by dexamethasone (Dex) of lipopolysacharide (LPS)-induced TNF-a production in whole blood samples. The amount of TNF-a produced by LPS-stimulation in the presence or absence of Dex was measured by ELISA, the degree of Dex inhibition of TNF-a production being an index of cell sensitivity to GCs. The assay was carried out using the Dyna-MIX™ TNF-a stimulation assay kits (Biosource Europe SA, Belgium) of the same batch number (Lot number) according to the manufacturer’s suggestions with minor modifications. Briefly, 25 µl of whole blood samples or TNF-a standards were incubated with 200 µl of stimulant solution (100 ng/ml LPS final concentration in RPMI) in the presence or absence of dexamethasone-21-phosphate (10-6 M final concentration in RPMI) (Dex, no D1159, Sigma Chemical Co., St. Louis MO, USA) in anti-TNF-a coated microtiter plates, for 3 h at 37oC. After incubation, washings, addition of anti-TNF-a horse-radish-peroxidase conjugate, chromogen and stop solution followed, as recommended by the manufacturer. Optical density was measured at 405 nm against a 650 nm blank filter on a CODA automated ELISA analyzer (Bio_Rad Laboratories, Richmond, CA, USA), and the results were calculated against a standard curve. The detection limit for TNF-a was 8 pg/ml, and the overall analysis intra-assay variability was 6% for the LPS-stimulated samples and 8 to 9% for the samples treated with Dex. All LPS-stimulated samples with or without Dex were measured in triplicate. The inter-assay variability of the TNF-a assay was 10%.
TNF-α assay
This assay was performed as described for the TNF-a stimulation assay (DynaMix™ TNF-a stimulation assay kit, Biosource Europe SA, Belgium), but 25 µl of whole blood was incubated with 200 µl of RPMI instead of stimulant solution.
Lymphocyte Glucocorticoid sensitivity index
The percent inhibition of LPS-induced TNF-a production in the presence of 10-5 M and 10-6 M DEX was taken as an index of sensitivity to GCs.
Cortisol assay
Serum cortisol was determined by radioimmunoassay (Coat-A-count, Cortisol RIA, Diagnostic Products Corporation, Los Angeles, CA, USA).
Individual feedback sensitivity of the HPA axis
As an index of the individuals’ feedback sensitivity of the HPA axis we used the degree of dexamethasone-induced suppression of cortisol levels, expressed as ƒF= (Fb-F post-dex)/Fb, where Fb means basal cortisol levels and F post-dex cortisol levels after the administration of 0.25mg dexamethasone.
RESULTS
As expected, there was a significant suppression of morning cortisol levels after the administration of 0.25mg of dexamethasone, mean baseline cortisol levels were 14.8µg/dl whereas mean post-DEX cortisol levels were 7.3µg/dl (p=0,001). As shown in figure 1, a considerable variability among the subjects was observed in ƒF, ranging from insignificant to almost 80% reduction reflecting differences among healthy individuals as to their feedback sensitivity of HPA axis.
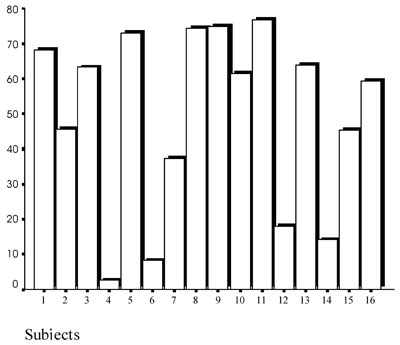
Figure 1. Distribution of ΔF* among the 16 individuals.
*ΔF= (Fb-F post-dex)/Fb
The LPS-induced TNF-a production by the individuals’ leukocytes was inhibited by dexamethasone in a dose-dependent manner. As shown in figure 2, a considerable inter-individual variation was also found.
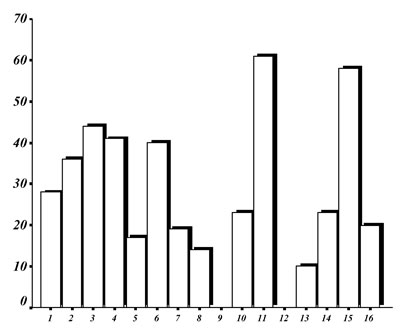
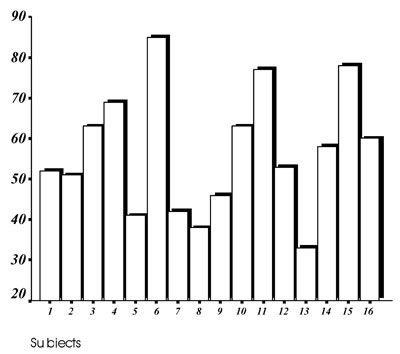
Figure 2.Distribution of the % inhibition of the LPS-induced TNF-a production by the individuals’ leukocytes by 10-6 M (upper panel) and 10-5 M (lower panel) dexamethasone.
As shown in figure 3, intercorrelations between the two markers of tissue GC sensitivity yielded no correlation between them. No significant differences between men and women were observed in respect to the degree of cortisol suppression or lymphocyte sensitivity (Table 1).
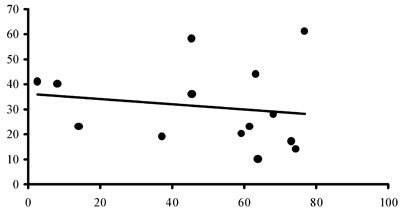
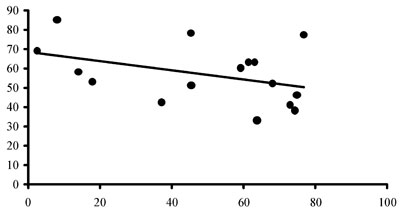
Figure 3.Scattergrams for correlation between ΔF and the % inhibition of the LPS-induced TNF-a production by the individuals’ leukocytes by 10-6 M (upper panel) and 10-5 M (lower panel) dexamethasone.
DISCUSSION
In the present study and in agreement with previous reports3-8, we demonstrated a wide variability in the tissue sensitivity to glucocorticoids among healthy subjects. Thus, in both GC targets assessed in this study, namely, cells of the peripheral immune system and the HPA axis, there was a wide variation in the magnitude of the responses observed. Furthermore, the GC sensitivity of the immune system of a given subject did not correlate with that observed with regard to HPA axis inhibition, indicating a tissue specificity of GC sensitivity in the same individual.
The correlation of GC sensitivity in different tissues of the same individual has been investigated in a limited number of reports with conflicting results. Previous studies including patients suffering from depression or asthma demonstrated a concordance in the GC sensitivities of the various functions tested. Thus, cortisol non suppression in patients with depression correlated positively with the lack of dexamethasone-induced inhibition of Con-A stimulated lymphocytes15. Also, steroid resistant asthma co-existed with a decreased sensitivity of skin blanching to exogenous beclomethasone application16. However, studies in normal volunteers, in good agreement with our findings, failed to demonstrate any correlation in the GC sensitivities of the target functions tested. Thus, in two recent studies no correlation was found between the lymphocyte glucocorticoid sensitivity and the cutaneous vasoconstrictor response to beclomethasone2,6. However, the use of two different types of steroids (dexamethasone and beclomethasone) raises questions as to whether or not the lack of correlation is due to variations in the tissue specificities or is simply the result of ligand-specific effects at different sites of action.
Our findings are similar to those recently reported by Ebrecht et al17 who found no correlation between the dexamethasone-sensitivity of the immune system and the feedback sensitivity to dexamethasone of the HPA axis. In the study of Ebrecht et al., GC lymphocyte sensitivity was measured by calculating the IC50 values of dexamethasone concentrations needed to block IL-6 and TNF-a production. However, IC50 values demonstrate relatively smaller variation between GC sensitive and resistant subjects7. In our study, we used a calculation of GC sensitivity corresponding to the Imax value, which varies more widely between subjects with different degrees of GC sensitivity7. Also, by using an even lower dose of dexamethasone than the dose used by Ebrecht et al. and the more accurate serum instead of salivary cortisol measurements, we reached a greater degree of variation in the feedback sensitivity in our subjects4. Despite these modifications in our protocol, our findings were analogous to those of Ebrecht et al., indicating no correlation between immune GC sensitivity and the feedback sensitivity of the HPA axis. A caveat in our study is that the immune function was tested at almost maximum suppression while the HPA axis was only minimally suppressed. However, maximum suppression of the HPA axis invalidates its use as a marker of GC sensitivity of this particular target.
There are several clinical implications of these findings. As measurements of GC sensitivity are increasingly used to predict the therapeutic outcome of steroid administration in patients with various inflammatory disorders, it should be pointed out that only direct measurements of the immune GC sensitivity should be used. Inflammation per se is a major determinant of immune sensitivity to glucocorticoids18,19. Indirect measurements in tissues of non-immune origin, like the in vivo assessment of the feedback sensitivity or the in vitro use of skin fibroblasts, may not be representative of the steroid actions on immune cells. On the other hand, the degree of immune GC sensitivity does not seem to predict the extent of steroid side effects. Thus, ideally for an individualized approach of steroid therapy, additional measurements of GC sensitivities in steroid-vulnerable tissues will be required for optimal management.
In conclusion, the bulk of current evidence suggests that GC sensitivity is tissue specific for a given individual. However, the exact mechanism of this phenomenon is still unknown. There might be several explanations for the tissue specificity of the GC action such as a variable expression level of the GR, a differential usage of the GR promoter and a differential expression of the various isoforms of the GR in various tissues1,6,20,21. Also, alterations in the steroid metabolizing enzymes or other interacting transcription factors (AP-1) which modulate hormone access to their receptors may also participate in conferring tissue specificity22,23. Moreover, it should be taken into account that GC-sensitivity is a dynamic rather than a static phenomenon. For example, GC sensitivity in various tissues demonstrates a seasonal and even a diurnal variation3,11. Also, various types of stressors affect GC sensitivity in several target tissues24-27. The role of an altered GR sequence in the observed GC sensitivity is also of high current research interest3,28,29. Additional studies focusing on each one of these possibilities are therefore warranted in order to elucidate the exact mechanism(s) involved in the differential GC tissue responsiveness.
REFERENCES
1. Bamberger CM, Schulte HM, Chrousos GP, 1996 Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocrine Reviews 17: 245-261.
2. Walker BR, Best R, Shackleton CHL, Padfield PL, Edwards CRW, 1996 Increased vasoconstrictor sensitivity to glucocorticoids in essential hypertension. Hypertension 27: 190-196.
3. Walker BR, Best R, Noon JP, Watt GCM, Webb DJ, 1997 Seasonal variation in glucocorticoid activity in healthy men. J Clin Endocrinol Metab 82: 4015_ 4019.
4. Huizenga NA, Kooper JW, de Lange P, et al, 1998 Interperson variability but intraperson stability of baseline cortisol concentrations, and its relation to feedback sensitivity of the hypothalamic-pituitary-adrenal axis to a low dose of dexamethasone in elderly individuals. J Clin Endocrinol Metab 83: 47-54.
5. Huizenga NA, Koper JW, de Lange P, 1998 A polymorphism in the glucocorticoid receptor gene may be associated with an increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab 83: 144 _151.
6. Panarelli M, Holloway CD, Fraser R, Connell JM, Ingram MC, Anderson NH, Kenyon CJ, 1998 Glucocorticoid receptor polymorphism, skin vasoconstriction, and other metabolic intermediate phenotypes in normal human subjects. J Clin Endocrinol Metab 83: 1846 _1852.
7. Hearing SD, Norman M, Smyth C, Foy C, Dayan CM, 1999 Wide variation in lymphocyte steroid sensitivity among healthy human volunteers. J Clin Endocrinol Metab 84: 4149-4154.
8. Corrigan CJ, Brown PH, Barnes NC, 1991 Glucocorticoid resistance in chronic asthma: glucocorticoid pharmacokinetics, glucocorticoid receptor characteristics, and inhibition of peripheral blood T cell proliferation by glucocorticoids in vitro. Am Rev Respir Dis 144: 1016 _1025.
9. Corrigan CJ, Bungre JK, Assoufi B, Cooper AE, Seddon H, Kay AB, 1996 Glucocorticoid resistant asthma: T-lymphocyte steroid metabolism and sensitivity to glucocorticoids and immunosuppressive agents. Eur Respir J 9: 2077_2086.
10. Sher ER, Leung DY, Surs W, Kam JC, Zieg G, Kamada AK, Szefler SJ, 1994 Steroid-resistant asthma. Cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest 93: 33-39.
11. Gratsias Y, Moutsatsou P, Chrysanthopoulou G, Tsagarakis S, Thalassinos N, Sekeris CE, 2000 Diurnal changes in glucocorticoid sensitivity in human peripheral blood samples. Steroids 65: 851_856.
12. DeRijk R, Michelson D, Karp B, 1997 Exercise and circadian rhythm-induced variations in plasma cortisol differentially regulate interleukin-1 b (IL-1 b), IL-6, and tumor necrosis factor- a (TNFa) production in humans: high sensitivity of TNFa and resistance of IL-6. J Clin Endocrinol Metab 82: 2182_2191.
13. Panarelli M, Holloway CD, Mulatero P, Fraser R, Kenyon CJ, 1994 Inhibition of lysozyme synthesis by dexamethasone in human mononuclear leukocytes: an index of glucocorticoid sensitivity. J Clin Endocrinol Metab 78: 872-877.
14. Haczku A, Alexander A, Brown P, 1994 The effect of dexamethasone, cyclosporine, and rapamycin on T-lymphocyte proliferation in vitro: comparison of cells from patients with glucocorticoid-sensitive and glucocorticoid-resistant chronic asthma. J Allergy Clin Immunol 93: 510 _519.
15. Lowy MT, Reder AT, Gormley GJ, Meltzer HY, 1988 Comparison of in vivo and in vitro glucocorticoid sensitivity in depression: relationship to the dexamethasone suppression test. Biol Psychiatry 24: 619 _630.
16. Brown PH, Teelucksingh S, Matusiewicz SP, Greening AP, Crompton GK, Edwards CR, 1991 Cutaneous vasoconstrictor response to glucocorticoids in asthma. Lancet 337: 576 _580.
17. Ebrecht M, Buske-Kirschbaum A, Hellhammer D, Kern S, Rohleder N, Walker B, Kirschbaum C, 2000 Tissue specificity of glucocorticoid sensitivity in healthy adults. J Clin Endocrinol Metab 85: 3733-3739.
18. Meduri GU, Tolley EA, Chrousos GP, Stentz F, 2002 Prolonged methylprednisolone treatment suppresses systemic inflammation in patients with unresolving acute respiratory distress syndrome: evidence for inadequate endogenous glucocorticoid secretion and inflammation-induced immune cell resistance to glucocorticoids. Am J Respir Crit Care Med 165: 983-91.
19. Webster JC, Oakley RH, Jewell CM, Cidlowski JA, 2001 Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative b isoform: A mechanism for the generation of glucocorticoid resistance. PNAS 98: 6865_6870.
20. Bamberger CM, Bamberger A-M, de Castro M, Chrousos GP, 1995 Glucocorticoid receptor ™, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95: 2435_2441.
21. Yudt MR, Cidlowski JA, 2002 The glucocorticoid receptor: coding a diversity of proteins and responses through a single gene. Mol Endocrinol 16: 1719-26.
22. Adcock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ, 1995 Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. Exp Med 182: 1951_1958.
23. Walker BR, 1994 Organ-specific actions of 11 beta-hydroxysteroid dehydrogenase in humans: implications for the pathophysiology of hypertension. Steroids 59: 84-89.
24. Derijk RH, Petrides J, Deuster P, Gold PW, Sternberg EM, 1996 Changes in glucocorticoid sensitivity of peripheral blood lymphocytes after strenuous exercise in humans. J Clin Endocrinol Metab 81: 228-235.
25. Van Rijen E, Harvey RA, Barton RN, Rose JG, Horan MA, 1998 Sensitivity of mononuclear leucocytes to glucocorticoids in elderly hip-fracture patients resistant to suppression of plasma cortisol by dexamethasone. Eur J Endocrinol 138: 659 _666.
26. Rohleder N, Kudielka BM, Hellhammer DH, Wolf JM, Kirschbaum C, 2002 Age and sex steroid-related changes in glucocrticoid sensitivity of pro-inflammatory cytokine production after psychological stress. J Neuroimmunol 126: 69-77.
27. Molijn GJ, Spek JJ, van Uffelen JC, de Jong FH, Brinkmann AO, Bruining HA, 1995 Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J Clin Endocrinol Metab 80: 1799-1803.
28. Moutsatsou P, Tsolakidou A, Trikkas G, Troungos C, Sekeris CE, 2000 Glucocorticoid receptor alpha and beta isoforms are not mutated in bipolar affective disorder. Molecular Psychiatry 5: 196-202.
29. Koper JW, Stolk RP, de Lange P, et al, 1997 Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Human Genet 99: 663-8.
Address correspondence and requests for reprints to:
S. Tsagarakis MD, PhD, Dept of Endocrinology,
Evangelismos Hospital, 106 76 Athens, Greece, E-mail: stsagara@otenet.gr
Received 05-08-02, Revised 15-08-02, Accepted 20-09-02