Address Correspondence to:
Edward M. Brown, M.D., Endocrine-Hypertension Division, Brigham and Women’s Hospital, 221 Longwood Ave., Boston, MA 02115, Phone 617-732-5661, Fax 617-732-5764, E-mail:embrown@rics.bwh.harvard.edu
Received 07-11-2001, Accepted 10-12-2001
Abstract
The system that regulates extracellular calcium (Ca2+o) homeostasis maintains a nearly constant level of Ca2+o so as to ensure the availability of calcium for its numerous intra- and extracellular roles. The molecular cloning and characterization of a G protein-coupled, Ca2+o-sensing receptor has elucidated the mechanism through which parathyroid cells and other cell types involved in calcium homeostasis sense Ca2+o and initiate the homeostatic responses that maintain Ca2+o at its normal level. The identification of the CaR has also proven unequivocally that extracellular calcium ions serve in an informational capacity. Furthermore, the identification of inherited human disorders resulting from inactivating and activating mutations of the CaR that produce hyper- and hypocalcemia, respectively, has provided physiological proof of the essential role of the CaR in mineral ion metabolism. Finally, selective activators of the CaR, so-called calcimimetics, are in clinical trials for the treatment of primary and uremic hyperparathyroidism and will likely provide the first truly effective medical treatment of hyperparathyroidism. CaR antagonists (calcilytics) may also prove to be of clinical utility in settings where inhibition of the receptor would be desirable.
Keywords: Calcium-Sensing Reseptor, hypocalcemia, Hypercalcemia
Essentially all physiological processes utilize intra- and/or extracellular calcium (Ca2+) ions in some fashion. The intracellular free calcium concentration (Ca2+i) serves as a key intracellular second messenger and enzymic cofactor, regulating numerous critical cellular functions (i.e., muscular contraction, hormonal secretion, glycogen metabolism, cellular differentiation, proliferation and motility)1. The resting level of Ca2+i, is 100 nanomolar (nM), which is about ten thousand-fold lower than the extracellular ionized calcium concentration (Ca2+o) (1.3 mM). Ca2+i can rise rapidly to levels of 1 uM or higher upon cellular activation by hormones and other factors as a result of the release of Ca2+ from intracellular stores2 and/or uptake of extracellular calcium1. In contrast to Ca2+i, the level of Ca2+o in the blood remains nearly constant under normal circumstances over the course of a day or even a lifetime, varying from its mean value by only a few percent3. Ca2+o ultimately provides the source of all intracellular calcium, and Ca2+o is also essential for numerous key biological functions in the extracellular space, such as clotting of the blood, maintenance of skeletal integrity, intercellular adhesion and regulation of neuromuscular excitability-further emphasizing the need for a mechanism maintaining near constancy of Ca2+o3.< Overview of the Ca2+o homeostatic systemFree-living terrestrial organisms have evolved a complex homeostatic mechanism that is responsible for the near invariance of Ca2+o in the blood (Figure 1) [for review, see3]. It is generally thought of as having two principal elements: The first are Ca2+o-sensing cells that secrete either Ca2+o-elevating [parathyroid hormone (PTH) or 1,25-dihydroxyvitamin D] or Ca2+o-lowering [calcitonin (CT)] hormones into the systemic circulation. These cells include the PTH-secreting chief cells of the parathyroid glands, the CT-secreting C-cells of the thyroid and the 1,25 -dihydroxyvitamin D3 [1,25 (OH) 2D3]- synthesizing cells of the renal proximal tubule. Elevating Ca2+o inhibits PTH secretion, decreases the production of 1,25 (OH) 2D3 directly4 and indirectly (i.e., by inhibiting PTH release) and increases the secretion of CT. PTH and 1,25 (OH) 2D3 are the major Ca2+o-regulating hormones in humans, while calcitonin has little importance under normal physiological circumstances, although it may be useful in the therapy of certain disorders (e.g., Paget’s disease). In response to hypocalcemia, increased circulating levels of PTH and 1,25 (OH)2D3 act on their various effector tissues, principally intestine, kidney and bone, and-by altering the transport of calcium ions into or out of the extracellular fluid–normalize Ca2+o (Figure 1) [for review, see3].
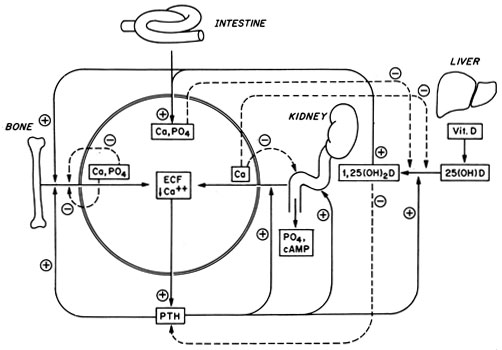
Figure 1. Schematic diagram of the homeostatic system via which Ca2+o is maintained nearly constant in the blood and extracellular fluids. The solid arrows and lines show the actions of parathyroid hormone (PTH) and 1,25-hydroxyvitamin D3 [1,25(OH)2D3] on their target tissues; the dotted arrows and lines show direct actions of Ca2+o and phosphate ions on the same tissues. Other abbreviations: Ca2+, calcium; P04, phosphate; ECF, extracellular fluid; 25(OH)D, 25-hydroxyvitamin D; minus signs show inhibitory effects, and plus signs indicate stimulatory actions. Reproduced with permission from Brown EM, Pollak M, and Hebert SC. Cloning and characterization of extracellular Ca2+-sensing receptors from parathyroid and kidney: Molecular physiology and pathophysiology of Ca2+-sensing. The Endocrinologist. 1994;4;419-426.
Thus the capacity of the cells secreting Ca2+o-regulating hormones to recognize and respond to small, physiologically relevant changes in Ca2+o is a crucial element in Ca2+o homeostasis. Changes in Ca2+o also, however, have direct actions on the cells that translocate Ca2+o into and out of the extracellular fluid–inhibiting, for example, distal tubular reabsorption of calcium5, promoting bone formation6,7 and inhibiting bone resorption8,9. In effect, therefore, Ca2+o not only regulates the secretion of the classical Ca2+o-regulating hormones but also modulates their actions at the level of their target tissues and by directly regulating the latter. As described in more detail below, the cloning of a G protein-coupled receptor that mediates a number of these direct actions of Ca2+o-the Ca2+o-sensing receptor (abbreviated as CaR or CaSR)-demonstrates that Ca2+o, acting via the CaR, can itself be viewed as a Ca2+o-regulating “hormone”3. It does so both at systemic (i.e., by regulating PTH secretion) and local levels (i.e., by modulating renal tubular reabsorption of calcium)10. Indeed, Ca2+o can be viewed as the body’s principal Ca2+o-lowering hormone.
Role of the G protein-coupled extracellular Ca2+-sensing receptor (CaR) in Ca2+o-sensing by parathyroid and diverse other cells.
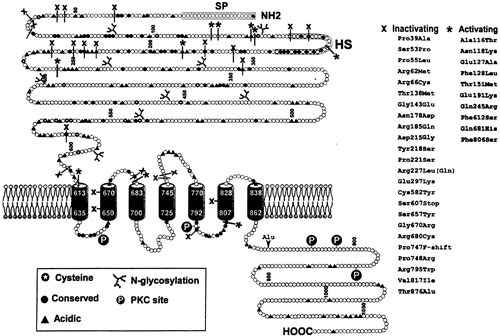
Figure 2. Predicted topology of the Ca2+o-sensing receptor (CaR) cloned from human parathyroid. Abbreviations are: SP, signal peptide; HS, hydrophobic segment. Also indicated are some of the missense and nonsense mutations that cause either familial hypocalciuric hypercalcemia (FHH) or autosomal dominant hypocalcemia. Numerous additional ones are now known. These are shown using the three letter amino acid code. The normal amino acid is shown before and the mutated amino acid is indicated following the numbers of the relevant codons. Reproduced in modified form with permission from Brown EM, Bai M, Pollak M. Familial benign hypocalciuric hypercalcemia and other syndromes of altered responsiveness to extracellular calcium. In Metabolic Bone Diseases., Krane SM, Avioli LV, eds, third ed., Academic Press: San Diego, CA; 1997; 479-499.
Cloning and characteristics of the CaR
The use of expression cloning in Xenopus laevis oocytes enabled the isolation of a cDNA clone encoding a full length, biologically active CaR from a cDNA library prepared from bovine parathyroid11. The use of hybridization-based screening techniques then permitted the cloning of additional, highly homologous CaRs from human parathyroid (Figure 2) and kidney, as well as from rat kidney, C-cell, and brain [for review, see3. These various CaRs are all activated not only by high Ca2+o but also by Mg2+o and diverse inorganic [e.g., the trivalent cation, gadolinium (Gd3+)] and organic polycations (i.e., spermine and neomycin)11. Ca2+o, and perhaps Mg2+o, are thought to be the physiologically relevant agonists of the receptor in vivo, although it is possible that spermine, when present in certain locations within the body at sufficiently high levels, could also modulate the CaR12.
The CaR has three major structural features that can be predicted from its deduced amino acid sequence (Figure 2): 1) A large (~600 amino acid) amino-terminal extracellular domain (ECD) that plays an important role in sensing Ca2+o, 2) seven membrane-spanning helices that are the signature of the GPCRs and 3) an intracellular C-terminal tail of some 200 amino acid residues13. The CaR’s C-tail as well as its intracellular loops likely play important roles in coupling the receptor to its intracellular effector systems, in a G protein-dependent manner14. The latter include CaR-mediated activation of phospholipases C, D and A215 as well as various mitogen-activated protein kinases (MAPK)16,17, and inhibition of adenylate cyclase18.
Tissue distribution of the CaR
The CaR is expressed at the highest levels in parathyroid cells, C-cells19,20 as well as in the distal tubule of the kidney21, and its functional roles in these tissues is discussed below (for review, see3). The receptor is also expressed at lower levels in other nephron segments (e.g., the proximal tubule and distal collecting duct)21, along the entire length of the gastrointestinal tract22, as well as within growth plate cartilage23, osteoblastic cell lines24 and osteoblasts within sections of bone23 (although some investigators have not detected the receptor in osteoblastic cells25). It is also expressed in cells of the monocyte/macrophage lineage that could potentially serve as osteoclast precursors26. There is conflicting evidence as to whether it is present in osteoclasts or if Ca2+o-sensing in this cell type as well as in osteoblasts involves the CaR or some other molecular mechanism, as will be discussed in more detail later (for review, see3); its physiological relevance in the GI tract likewise remain uncertain.
The receptor is also expressed, however, in numerous tissues that are almost certainly uninvolved in Ca2+o homeostasis (for review, see3). For instance, in the intestine, the CaR is expressed in the surface cells of the gastric mucosa and widely throughout the enteric nervous system. Within the kidney, the CaR is present on the apical membrane of the inner medullary collecting duct, where it probably mediates the known inhibitory action of elevated Ca2+o on vasopressin-stimulated water reabsorption (see below)27. In bone marrow, the CaR is expressed in several hematopoietic lineages, particularly red blood cell precursors, megakaryocytes and monocytes. Other _non-homeostatic_ tissues that express the receptor include the epithelial cells of the breast ducts, keratinocytes, pancreas, various cell types within the central nervous system (e.g., hippocampal pyramidal, cerebellar and numerous other neurons as well as oligodendrocytes), lens epithelial cells and numerous others. Clearly a great deal remains to be learned about the receptor’s roles in these tissues, although it may respond to local changes in Ca2+o in ways that enable it to participate in their normal differentiated functions.
Control of parathyroid function and CT secretion by the CaR
Strong evidence supports the CaR’s role in mediating of the inhibitory actions of Ca2+o on PTH release and parathyroid cellular proliferation. The CaR’s importance in controlling PTH secretion is supported by the abnormalities in Ca2+o-regulated PTH release that are present in inherited human diseases with inactivating or activating mutations28 in the receptor and mouse models in which the CaR has been “knocked out”29. As described in more detail below, in human disorders in which the receptor has reduced activity as well as in mice with targeted disruption of one allele of the CaR gene, there is impaired inhibition of PTH secretion at any given level of Ca2+o. In contrast, in patients with CaRs that are overly sensitive to activation by Ca2+o due to the presence of activating mutations, there is a greater than normal suppression of hormonal secretion as a function of Ca2+o. In both humans who are homozygous for inactivating mutations of the CaR or in mice homozygous for knockout of the CaR gene, there is not only markedly abnormal Ca2+o-regulated PTH release but also substantial parathyroid gland hyperplasia28,29, strongly supporting the CaR’s role in tonically inhibiting parathyroid cellular growth. The CaR may also mediate other known actions of Ca2+o on parathyroid function, including inhibition of PTH gene expression30, although additional studies are needed in this regard. Despite intensive study over several decades the major intracellular mediators of the CaR’s actions on PTH secretion and other aspects of parathyroid function remain uncertain31. As noted above, the CaR is expressed at high levels in the C-cells of the thyroid gland19,20. Available evidence supports its involvement in activating signal transduction pathways that increase Ca2+i and stimulate CT secretion unlike the parathyroid cell in which CaR-induced increases in Ca2+i are associated with inhibition of PTH secretion32.
Control of renal function by the CaR
Several lines of evidence suggest that the CaR plays a key role in controlling Ca2+ and Mg2+ reabsorption in the renal distal tubule and represents the molecular mechanism for the direct inhibitory actions of Ca2+o and Mg2+o on divalent cation reabsorption in this portion of the nephron33. The CaR is expressed at high levels on the basolateral surface of the epithelial cells of the cortical thick ascending limb (CTAL) of the nephron21, where, as in parathyroid cells, it appears to couple to activation of PLA2 and inhibition of adenylate cyclase (for review, see3). The inhibitory, presumably CaR-mediated action of elevated Ca2+o on divalent cation reabsorption in CTAL is thought to result from a reduction in the reabsorption of sodium chloride by the NaK2Cl cotransporter that is coupled, in turn, to a concomitant decrease in tubular reabsorption of Ca2+ and Mg2+ via the paracellular pathway33. The CaR, in effect, acts like a loop diuretic, although it inhibits the cotransporter indirectly rather than directly as in the case with the diuretics33. This direct, local action of Ca2+o on tubular function likely provides a mechanism for local “autoregulation” of divalent cation handling by the CTAL in addition to the modulating the known stimulatory action of circulating PTH on this parameter (e.g., Figure 1).
In addition to their abnormal Ca2+o-regulated PTH secretion, patients with inactivating mutations of the CaR also exhibit overly avid renal tubular Ca2+ and Mg2+ reabsorption34. The latter presumably results from a reduced number of normally functioning CaRs in the CTAL, leading to “resistance” to the usual action of hypercalcemia to promote calciuria. Interestingly, the only maneuver that substantially increases urinary calcium excretion in these patients is administering the loop diuretic, ethacrynic acid35. This observation adds additional indirect evidence that defective CaR-mediated inhibition of the NaK2Cl cotransporter participates in their excessive tubular reabsorption of divalent cations. The CaR is also expressed in the distal convoluted tubule, another important site where PTH increases distal tubular reabsorption of calcium, but its functional role in this nephron segment requires additional investigation. It should be possible to use the mouse model with targeted disruption of the CaR to investigate further the role of the CaR in regulating divalent cation handling as well as other aspects of renal function.
Does the CaR regulate the functions of intestinal and bone cells?
The CaR’s presence in the epithelial cells of the small and large intestines that are involved in calcium absorption suggests that it could potentially be involved in regulating this process22. Its presence in the enteric nervous system could also implicate it in controlling intestinal secretomotor functions22. However, there is no direct evidence that the receptor regulates these functions, and additional studies are needed.
Ca2+o is known to stimulate various aspects of osteoblastic function, such as proliferation, chemotaxis, and the secretion of growth factors as well as bone formation per se in organ culture (for review, see24). Ca2+o also inhibits various aspects of osteoclastic function9. Whether the receptor mediates the functions of these bone cells, however, remains an open question. Some investigators have found the CaR to be expressed in osteoblastic cell lines, bone marrow stromal cells or in bone sections and to regulate some aspects of osteoblast function9,23, but others have not been able to demonstrate the receptor’s presence in cells of the osteoblastic lineage25. Furthermore, the latter investigators found that osteo-blastic cells from CaR knockout mice could still respond to changes in Ca2+o36. There is also controversy as to whether the CaR is in osteoclasts and/or their precursors and mediates known actions of Ca2+o on cells of the osteoclast lineage (for review, see24). The CaR is expressed at readily detectable levels in blood monocytes, which include cells that can serve as osteoclast precursors, and mediates the chemotactic response of these cells to elevated levels of Ca2+o26. Some but not all investigators have documented CaR expression in mature, multinucleated osteoclasts24,37. Again, however, more direct evidence is needed, particularly in view of the fact that there are pharmacological differences between the putative Ca2+o-sensors/receptors in osteoblasts and osteoclasts and the CaR9. Nevertheless, the capacity of bone cells to sense Ca2+o likely enables direct, local actions of Ca2+o on the functions of these cells that contribute additional homeostatic control to that afforded by more classical calciotropic hormone (e.g., PTH and 1,25 (OH) 2D3)7,24.
Roles of the CaR in integrating mineral ion homeostasis with other homeostatic systems
Hypercalcemic patients can manifest defective maximal urinary concentrating ability and, on occasion, overt nephrogenic diabetes insipidus10,28. The presence of the CaR in nephron segments that participate in urinary concentration (e.g., the inner medullary collecting duct) has provided novel insights into how high Ca2+o may regulate the renal concentrating mechanism27. Perfusion of the lumen of IMCD tubules from rat kidney with high Ca2+o reversibly inhibits vasopressin-evoked transepithelial water flow. In addition, the CaR is present in the same apical endosomes in cells of the IMCD that contain the vasopressin-regulated water channel, aquaporin-227. Therefore, the CaR could diminish vasopressin-activated water flow in the IMCD by decreasing the availability and/or the activity of the aquaporin-2 water channels in the apical membrane. CaR-induced reduction in NaCl reabsorption in the loop of Henle would also act to reduce the medullary countercurrent gradient, which would further decrease maximal urinary concentrating ability in hypercalcemic individuals. Of interest in this regard, patients with FHH concentrate their urine normally38, while those who harbor activating mutations of the CaR can develop symptoms of defective urinary concentration at normal or even subnormal blood calcium concentrations39, presumably because the former are resistant and the latter excessively sensitive to the actions of elevated Ca2+o on urinary concentration.
Thus it is possible that the CaR may provide a means of integrating divalent cation and water handling by the kidney in order to allow appropriate “trade-offs” in the regulation of these aspects of renal function that are appropriate for specific physiological circumstances. For instance, in circumstances when a systemic calcium load must be disposed of, coordinating the CaR-induced increase in urinary Ca2+ concentration owing to reduced calcium reabsorption in the CTAL with the associated decrease in maximal urinary concentrating capacity would diminish the maximal level of luminal Ca2+o in the IMCD. This could reduce the risk of Ca2+ stone formation10. The CaR is also expressed at high levels in the subfornical organ (SFO), an important thirst center in the hypothalamus40. High Ca2+o-evoked, CaR-mediated thirst and increased drinking could prevent dehydration otherwise resulting from renal free water loss owing to CaR-mediated renal resistance to vasopressin. Thus there may be multiple levels of integration of the homeo-static control systems for calcium and water metabolism, which may optimize the ability of free-living terrestrial organisms to adapt to their intermittent access to dietary calcium and water.
Recent studies have also shown that the CaR is activated by various amino acids, particularly aromatic amino acids, in the presence but not in the absence of physiologically relevant levels of Ca2+o (i.e., 1 mM)41. The presence of a mixture of amino acids emulating that present in the blood can sensitize the receptor significantly to Ca2+o41. This “multimodal” sensing by the CaR of both Ca2+o and amino acids may contribute to some of the known but poorly understood interactions between protein and calcium metabolism. For instance, high protein intake induces hypercalciuria and reduced protein intake produces substantial increases in circulating PTH levels in normal individuals (for review, see42). Further studies of the CaR’s participation in integrating mineral ion and protein metabolism may provide novel insights into how coordination of these homeostatic processes could contribute to processes involving the need for both mineral ions and protein, such as skeletal and somatic growth.
DISORDERS OF CALCIUM HOMEOSTASIS ARISING FROM ABNORMALITIES IN THE CAR
Disorders with generalized resistance to Ca2+o
The availability of the cloned CaR made it possible to search for disorders of mineral ion homeostasis resulting from abnormal structure and/or function of the CaR. Earlier studies had suggested that the autosomal dominant hypercalcemic disorder, familial hypocalciuric hypercalcemia (FHH), exhibited features consistent with generalized Ca2+o- “resistance”34,43. Despite their hypercalcemia, patients with FHH exhibit few, if any, of the usual symptoms, signs and complications of hypercalcemia. In addition, patients with FHH exhibit signs of resistance to the usual actions of hypercalcemia on parathyroid and kidney34,44: (1) Their serum PTH levels are inappropriately normal or mildly elevated in the face of hypercalcemia, consistent with “resistance” of the parathyroid to Ca2+o. Indeed, they exhibit an increase in the “set-point” for Ca2+o-regulated PTH release (the level of Ca2+o half-maximally suppressing circulating PTH levels)45,46. (2) They exhibit inappropriately normal or even low levels of renal tubular Ca2+ reabsorption given their hypercalcemia34,43. This abnormality in renal tubular Ca2+ handling persists even following complete parathyroidectomy, demonstrating that it is an intrinsic renal tubular defect and not the result of impaired high Ca2+o-induced suppression of PTH release35,47. (3) Finally, patients with FHH concentrate their urine normally in spite of their hypercalcemia, suggesting resistance to the normal inhibitory effect of hypercalcemia on the urinary concentrating mechanism38.
Another feature of FHH distinguishing it from PHPT is the unusual clinical course following parathyroidectomy. Unlike patients with PHPT, in whom parathyroidectomy usually produces long term cure of the hypercalcemia, performing anything less than total parathyroidectomy in persons with FHH is followed within a matter of days to weeks by recurrence of hypercalcemia34. From a clinical perspective, therefore, it is important to differentiate FHH from PHPT. Not only are individuals with the former condition generally asymptomatic, so that parathyroid surgery is of no apparent benefit, but it also generally fails to produce long term remission of hypercalcemia in FHH without inducing hypoparathyroidism. The general consensus has evolved, therefore, that persons with FHH should not undergo parathyroidectomy34,43,48. In occasional families with FHH, however, there can be overt hypercalciuria and even renal stones49 or the hypercalcemia can be quite severe (e.g., 13-14 mg/dl)50, and surgical therapy in these patients could potentially be of benefit. No doubt the clinical spectrum of FHH will enlarge further as our capacity to make a molecular diagnosis (see below) identifies more families with this condition.
The most helpful clinical clues to the diagnosis of FHH are as follows: (1) The presence of generally mild to moderate hypercalcemia (and, sometimes, mild hypermagnesemia) with an autosomal dominant pattern of inheritance, (2) intact PTH levels within the normal range and (3) relative hypocalciuria34,43,51. A useful way to document the abnormal renal Ca2+ handling is to calculate the ratio of the urinary calcium clearance to that for creatinine (the calcium to creatinine clearance ratio)34. Most patients with FHH will exhibit a value of 0.01 or less, while about 80% of patients with PHPT will have a value above 0.01. The diagnosis of FHH can in most cases be documented on the basis of these clinical and biochemical features and does not require genetic analysis to identify the mutations in the CaR that have subsequently been proven to be the cause of the vast majority of cases of this disorder51.
The gene for FHH was shown to reside on the long arm of chromosome 3 in 199252, although in rare families a biochemically similar disorder was also found to be linked to two different loci on chromosome 1953,54, documenting the genetic heterogeneity of this condition. Not long after the cloning of the CaR, three families with this condition were shown to harbor heterozygous missense mutations (i.e., a single base changes substituting a different amino acid for the one normally coded for) in the CaR’s coding region55. Subsequent studies by a number of laboratories have identified several dozen more missense mutations, occasional nonsense (e.g., mutation of a codon that encodes an amino acid to a stop codon) or other types of mutations in the CaR genes in FHH families, some of which are shown in Figure 2 [for review, see51]. When expressed in heterologous systems, such as human embryonic kidney (HEK) cells, these mutations produce varying degrees of inactivation of the CaR-varying from modest reductions in its apparent affinity for Ca2+o to its complete loss of function (Figure 3)56,57.
Therefore, reduction or loss of the normal function of one allele of the CaR gene produces a disorder with mild, generalized “resistance” of CaR-expressing tissues to Ca2+o. In the parathyroid, the resultant, modest (10-20%) increase in set-point is a major contributor to”resetting” the mineral ion homeostatic system to maintain a higher than normal Ca2+o. In the kidney, resistance to Ca2+o has two apparent consequences-(a) increasing the “set-point” of the kidney for high Ca2+o-evoked increases in urinary calcium excretion and (b) reducing the impairment of urinary concentration observed for a given degree of hypercalcemia [for review, see51]. This hypothesis is supported by a mouse model of FHH generated by targeted disruption of the CaR gene29. Mice heterozygous for CaR “knock-out” exhibit mild hypercalcemia (10.4 mg/dl), with normal PTH levels of PTH and relative hypocalciuria29. These mice appear otherwise well, resembling, therefore, patients with FHH. Mice homozygous for inactivation of the CaR gene, in contrast, have much more severe hypercalcemia (~15 mg/dl), accompanied by 5-10-fold elevations in PTH, severe bony demineralization and frequent fractures owing to severe hyperparathyroid bone disease29. They uniformly die within the first few weeks of life.
The clinical and biochemical findings in mice homozygous for knockout of the CaR resemble in many ways those of infants with neonatal severe hyperpara-thyroidism (NSHPT). Early descriptions of NSHPT emphasized these infants’ severe hypercalcemia (levels as high as 30.8 mg/dl have been reported) as a result of marked hyperparathyroidism due to four gland parathyroid hyperplasia48,58. These infants often had multiple fractures resulting from hyperparathyroid bone disease. These early studies generally recommended complete or subtotal parathyroidectomy as the treatment of choice for NSHPT, as the disorder was often fatal without surgical intervention in these early reports48,58. More recent reviews have documented a wider clinical spectrum for infants with NSHPT48. A substantial proportion of cases exhibit hyperparathyroid bone disease or bony demineralization accompanied by a milder degree of hypercalcemia (e.g., 12-14 mg/dl)48. In these cases, the condition is often self-limited with conservative medical therapy, and the clinical picture eventually reverts within several months to one that is not unlike FHH48. In fact, many of these infants have been found to be members of FHH kindreds48,58.
The cloning of the CaR and the demonstration that FHH is the result of heterozygous inactivating mutations of the CaR gene has subsequently elucidated the molecular basis for several cases of NSHPT. The most severe cases, usually with serum calcium concentrations of 14-16 mg/dl or higher, can represent FHH in its homozygous form (i.e. both alleles of the CaR gene harbor inactivating mutations)59 as a result of consanguineous marriages of two affected individuals. A more recently described infant with severe NSHPT was shown to be a compound heterozygote as a result the marriage of two individuals with different mutations in the CaR60. These cases of NSHPT due to homozygous or compound heterozygous CaR mutations usually require parathyroidectomy to avoid severe complications including death.
It is also apparent, however, that in many cases of NSHPT only one parent has clinically apparent FHH48,58. In some cases, the disorder may result, at least in part, from FHH mutations that exert “dominant negative” actions on the remaining normal CaR allele61. Because the active, cell surface form of the CaR is a dimer62, this “dominant negative” action likely results from the formation of CaR heterodimers -containing one wild type and one mutant CaR molecule- that have a reduced capacity to be activated over the normal range of Ca2+o. As a result, homodimeric CaRs containing two normal receptors would comprise on a purely statistical basis only 25% of the cell surface receptors and would represent the only species of the CaR on the cell surface with normal biological activity. Therefore, the “resistance” of parathyroid and kidney to Ca2+o in such cases would be greater than in the mouse model of FHH or in FHH families with mutations that are not expressed efficiently on the cell surface, because in the latter instances normal receptors (e.g., arising from the remaining normal allele of the CaR gene) would comprise closer to 50% of the cell surface CaR51. Indeed, in FHH families with mutations exerting dominant negative actions in vitro (e.g., R795W and R185Q), the hypercalcemia in affected family members is more severe (12.5 and 13-14 mg/dl, respectively) than in the majority of FHH families (~10.5-11.5 mg/dl)61. In addition, several affected infants in the family harboring the R185Q mutation presented with NSHPT50, even with only one affected parent. Similarly, a recently described infant with the same R185Q mutation61 proved to have arisen de novo, since both biological parents were unaffected. Several other infants have subsequently been described with de novo mutations causing NSHPT63. In these cases, it may be prudent to perform mutational analysis to confirm the diagnosis. In general, in most cases of NSHPT due to heterozygous CaR mutations, conservative medical therapy of the hypercalcemia will produce a successful clinical outcome if the serum calcium concentration is not excessively elevated (i.e., >15 mg/dl) and if parathyroidectomy is not deemed necessary on purely clinical grounds. Each case should be carefully evaluated on an individual basis, however, to minimize complications and to preclude the occasional fatal outcome in severely affected infants.
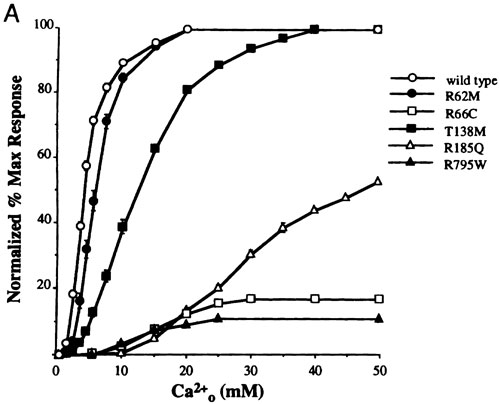
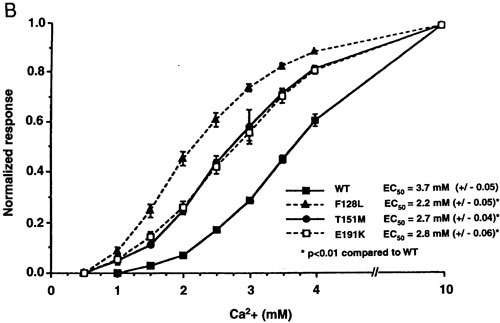
Figure 3. Expression in HEK 293 cells of CaRs harboring inactivating (A) and activating (B) mutations. The results show Ca2+o-induced increases in Ca2+i in HEK293 cells transiently transfected with the normal human CaR or CaRs bearing the mutations indicated by the single letter amino acid code. Note that inactivating mutations shift the dose response curve for Ca2+o to the right (reset the “calciostat” upward) and often reduce the maximal activity of the CaR, while inactivating mutations shift the curves to the left (reset the “calciostat” downward). Upper panel is reproduced with permission from Bai M, Quinn S, Trivedi S, Kifor O, Pearce SHS, Pollak MR, Krapcho K, Hebert SC, Brown EM. Expression and characterization of inactivating and activating mutations in the human Ca2+o-sensing receptor. J Biol Chem. 1996;271:19537-19545. Lower panel is reproduced with permission from Pearce SHS, Bai M, Quinn SJ, Kifor O, Brown EM, and Thakker RV. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest. 1996; 98:1860-1866.
Disorders resulting from “tissue-specific” Ca2+o-resistance
In primary and some cases of uremic secondary hyperparathyroidism (usually when there is overt hypercalcemia) -in contrast to FHH and NSHPT where there is generalized resistance to Ca2+o- the resistance to Ca2+o is present solely in the pathological parathyroid gland(s) [for review see51], while all other tissues in the body presumably have normal sensitivity to Ca2+o. Parathyroid cells prepared from pathological parathyroid tissue of patients with primary and severe uremic hyperparathyroidism have ~20-30% increases in their set-points for Ca2+o-regulated PTH release51, which is slightly higher than the elevation in set-point in FHH. Several studies have recently shown ~50% reductions in the levels of expression of CaR mRNA and/or protein in such pathological parathyroid glands relative to normal parathyroid tissue [for review, see51], without any mutations in the CaR’s coding region64. This reduced expression of the CaR may result from reduced activity of one of the two functional promoters of the CaR gene65. Thus the resistance of the parathyroid glands to Ca2+o in hyperparathyroidism as well as in FHH and NSHPT have in common a reduced complement of normal CaRs on the parathyroid cell surface.
A disorder resulting from generalized over responsiveness to Ca2+o
Because activating mutations in other GPCRs can cause disease66, such as hyperthyroidism caused by “activated” mutant TSH receptors67, it seemed possible that a disorder might exist that was the mirror image of FHH in other words, “familial hypercalciuric hypocalcemia”. Indeed, screening of families with autosomal dominant hypocalcemia for mutations in the CaR has revealed well over a dozen families harboring activating mutations39,68,69 (for review, see28). Individuals with activating mutations of the CaR may be asymptomatic, but they not infrequently exhibit symptoms present in other hypocalcemic disorders, including carpopedal spasm and/or seizures, particularly in those with more severe hypocalcemia. Affected individuals manifest mild to moderate and, in some cases, severe (5-6 mg/dl) hypocalcemia accompanied by normal to frankly elevated levels of serum phosphorus, and frankly low or low-normal levels of PTH. In the untreated state, these patients tend to have higher rates of urinary calcium excretion than do patients with hypoparathyroidism of other causes69, which can increase dramatically during treatment with calcium and vitamin D supplementation39. Some families also have hypomagnesemia39, although serum magnesium concentrations have not been reported in many families. De novo activating CaR mutations have also been described as a cause of sporadic hypocalcemia, emphasizing the importance of considering this diagnosis even in the absence of familial involvement69.
Expression of mutant CaRs engineered to contain activating mutations has revealed increases in the apparent affinities of these receptors for Ca2+o39 (Figure 3). Most of these mutations reside within the CaR’s ECD, emphasizing its importance in the mechanism of receptor activation following ligand binding, although some mutations are present within the CaR’s transmembrane domains, the most common location for naturally-occurring activating mutations of other GPCRs66. These activating mutations “reset” the calcium homeostatic system downward such that it defends a stable, albeit lower than normal level of Ca2+o. Thus, analogous to FHH, autosomal dominant hypocalcemia owing to activating CaR mutations is a disorder in which the “calciostat” is reset-upward in the case of FHH and downward with activating mutations. Nevertheless, autosomal dominant hypo-calcemia due to activating CaR mutations resembles hypoparathyroidism clinically in the sense that there is insufficient PTH to maintain normocalcemia.
Treatment of hypocalcemia resulting from activating mutations should be limited to symptomatic patients, because of their tendency to develop renal complications during treatment with vitamin D and calcium supplementation, which can produce severe hypercalciuria, nephrocalcinosis, renal stones and impaired renal function39. The hypercalciuria of patients with activating mutations in the untreated state as well as during treatment is almost certainly the result of “activated” CaRs in the kidney that reduce renal tubular Ca2+ reabsorption the opposite of the abnormality in renal Ca2+ handling observed in FHH. During treatment with vitamin D and calcium supplementation, therefore, urinary calcium excretion in addition to serum calcium concentration should be monitored carefully to avoid renal complications. The serum calcium concentration should be raised just enough to alleviate symptoms without producing excessive hypercalciuria. In some cases it may be necessary to use a thiazide diuretic to reduce urinary calcium excretion if symptoms persist in the presence of overt hypercalciuria during treatment with calcium and vitamin D supplementation.
In summary, it is important to consider the diagnosis of an activating CaR mutation in patients with what appears to be hypoparathyroidism but occurs in association with varying combinations of the following constellation of findings: (a) low-normal levels of serum PTH in the untreated state, (b) hypomagnesemia, (c) the occurrence of hypocalcemia in other family members with an autosomal dominant pattern of inheritance and (d) a tendency to hypercalciuria in the untreated state as well as marked hypercalciuria that is accompanied by impaired renal function, nephrolithiasis and/or nephrocalcinosis during treatment with vitamin D and calcium supplementation. In sporadic cases with these findings, mutational analysis may be appropriate to establish the diagnosis, although it is not available on a routine basis. Only symptomatic patients should be treated, and they should be monitored carefully during treatment with regular determinations of serum and urinary calcium concentrations to avoid renal complications.
CAR-BASED THERAPEUTICS
The CaR potentially represents an important therapeutic target for diseases in which the receptor is inappropriately over- or underactive70. To date CaR-based therapy has been directed principally at primary and uremic hyperparathyroidism71,72. Clinical trials are currently ongoing that are addressing the efficacy of the so-called “calcimimetic” CaR activators in the treatment of primary and uremic hyperparathyroidism. These agents produce rapid (within minutes) and substantial (>50%) decreases in circulating PTH, followed several hours later by reductions in serum calcium concentration at higher doses. When administered for several months calcimimetics can normalize serum calcium and PTH concentrations in most patients without promoting hypercalciuria, suggesting that they activate the CaR in the parathyroid to a greater extent than that in the kidney. In effect, the calcimimetics “reset” the elevated set-point of patholo-gical parathyroid glands toward normal73. CaR agonists will likely also be very useful for treating uremic hyperparathyroidism. Available data suggest that they lower the calcium-phosphate product as well as serum PTH levels, thereby providing an effective means of treating or mitigating the complications associated with abnormal mineral ion metabolism in renal failure72. CaR antagonists, so-called “calcilytics”, are available and are under investigation as a means of stimulating PTH secretion in an intermittent fashion, which might represent an alternative approach to the administration of exogenous PTH in the treatment of osteoporosis74.
SUMMARY AND CONCLUSIONS
Our understanding of the molecular basis for Ca2+o-sensing has increased greatly since the cloning of the G protein-coupled, Ca2+o-sensing receptor. The receptor plays a key role in maintaining the near constancy of Ca2+o through its actions the effects of Ca2+o on PTH secretion and renal calcium reabsorption. Ca2+o-sensing by the CaR or some other mechanism may also modulate the functions or intestinal and/or bone cells in ways that are physiologically relevant. The CaR also appears to participate in integrating Ca2+o homeostasis with other homeostatic systems (e.g., those regulating water and protein metabolism). The identification of inherited diseases of Ca2+o-sensing resulting from inactivating or activating mutations in the receptor has provided proof of the central role of the CaR in calcium homeostasis, as has the development of a mouse model with knockout of the CaR gene. Finally, CaR-based therapeutics that allosterically activate the receptor will likely provide the first truly effective medical treatment for primary and uremic secondary hyperparathyroidism and antagonists of the CaR also exhibit therapeutic promise.
The author is supported by NIH grants, DK48330, DK52005 and DK41415, and grants from the St. Giles Foundation and NPS Pharmaceuticals.
REFERENCES
1. Pietrobon D, Di Virgilio F, Pozzan T, 1990 Structural and functional aspects of calcium homeostasis in eukaryotic cells. Eur J Biochem 120: 599-622.
2. Meldolesi J, Pozzan T, 1998 The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci 23: 10-4.
3. Brown EM, MacLeod RJ, 2001 Extracellular Calcium Sensing and Extracellular Calcium Signaling. Physiol Rev 81: 239-297.
4. Weisinger JR, Favus MJ, Langman CB, Bushinsky D, 1989 Regulation of 1,25-dihydroxyvitamin D3 by calcium in the parathyroidectomized, parathyroid hormone-replete rat. J Bone Miner Res 4: 929-935.
5. Quamme GA, 1982 Effect of hypercalcemia on renal tubular handling of calcium and magnesium. Can J Physiol Pharmacol 60: 1275-80.
6. Raisz LG, Kream BE, 1983 Regulation of bone formation. N Engl J Med 309: 29-35.
7. Quarles LD, 1997 Cation-sensing receptors in bone: A novel paradigm for regulating bone remodeling? J Bone Miner Res 12: 1971-1974.
8. Raisz LG, Niemann I, 1969 Effect of phosphate, calcium, and magnesium on bone resorption and bone formation in tissue culture. Endocrinology 85: 446-452.
9. Zaidi M, Adebanjo OA, Moonga BS, Sun L, Huang CL, 1999 Emerging insights into the role of calcium ions in osteoclast regulation. J Bone Miner Res 14: 669-74.
10. Hebert SC, Brown EM, Harris HW, 1997 Role of the Ca(2+)-sensing receptor in divalent mineral ion homeo-stasis. J Exp Biol 200: 295-302.
11. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC, 1993 Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366: 575-80.
12. Quinn SJ, Ye CP, Diaz R, Kifor O, Bai M, Vassilev P, Brown E, 1997 The Ca2+-sensing receptor: a target for polyamines. Am J Physiol 273: C1315-23.
13. Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC, Nemeth EF, Fuller F, 1995 Molecular cloning and functional expression of human para-thyroid calcium receptor cDNAs. J Biol Chem 270: 12919-25.
14. Chang W, Chen TH, Pratt S, Shoback D, 2000 Amino acids in the second and third intracellular loops of the parathy-roid Ca2+)-sensing receptor mediate efficient coupling to phospholipase C. J Biol Chem 275: 19955-63.
15. Kifor O, Diaz R, Butters R, Brown EM, 1997 The Ca2+-sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR-transfected, human embryonic kidney (HEK293) cells. J Bone Miner Res 12: 715-25.
16. Kifor O, MacLeod RJ, Diaz R, Bai M, Yamaguchi T, Yao T, Kifor I, Brown EM, 2001 Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells. Am J Physiol Renal Physiol 280: F291-302.
17. McNeil SE, Hobson SA, Nipper V, Rodland KD, 1998 Functional calcium-sensing receptors in rat fibroblasts are required for activation of SRC kinase and mitogen-activated protein kinase in response to extracellular calcium. J Biol Chem 273: 1114-20.
18. Chang W, Pratt S, Chen TH, Nemeth E, Huang Z, Shoback D, 1998 Coupling of calcium receptors to inositol phosphate and cyclic AMP generation in mammalian cells and Xenopus laevis oocytes and immunodetection of receptor protein by region-specific antipeptide antisera. J Bone Miner Res 13: 570-80.
19. Garrett JE, Tamir H, Kifor O, Simin RT, Rogers KV, Mithal A, Gagel RF, Brown EM, 1995 Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology 136: 5202-11.
20. Freichel M, Zink-Lorenz A, Holloschi A, Hafner M, Flockerzi V, Raue F, 1996 Expression of a calcium-sensing receptor in a human medullary thyroid carcinoma cell line and its contribution to calcitonin secretion. Endocrinology 137: 3842-8.
21. Riccardi D, Hall AE, Chattopadhyay N, Xu JZ, Brown EM, Hebert SC, 1998 Localization of the extracellular Ca(2+)/polyvalent cation-sensing protein in rat kidney. Am J Physiol 274: F611-22.
22. Chattopadhyay N, Cheng I, Rogers K, Riccardi D, Hall A, Diaz R, Hebert SC, Soybel DI, Brown EM, 1998 Identification and localization of extracellular Ca(2+)-sensing receptor in rat intestine. Am J Physiol 274: G122-30.
23. Chang W, Tu C, Chen T-H, Komuves L, Oda Y, Pratt S, Miller S, Shoback D, 1999 Expression and signal transduction of calcium-sensing receptors in cartilage and bone. Endocrinology 140: 5883-93.
24. Yamaguchi T, Chattopadhyay N, Brown EM, 2000 G protein-coupled extracellular Ca(2+) (Ca(2+)o-sensing receptor (CaR): roles in cell signaling and control of diverse cellular functions. Adv Pharmacol 47: 209-53.
25. Pi M, Hinson TK, Quarles L, 1999 Failure to detect the extracellular calcium-sensing receptor (CasR) in human osteoblast cell lines. J Bone Miner Res 14: 1310-9.
26. Olszak IT, Poznansky MC, Evans RH, Olson D, Kos C, Pollak MR, Brown EM, Scadden DT, 2000 Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J Clin Invest 105: 1299-305.
27. Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW, 1997 Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest 99: 1399-405.
28. Brown EM, 1999 Physiology and pathophysiology of the extracellular calcium-sensing receptor. Am J Med 106: 238-53.
29. Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE, 1995 A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet 11: 389-94.
30. Garrett J, Steffey M, Nemeth E, 1995 The calcium receptor agonist R-568 suppresses PTH mRNA levels in cultured bovine parathyroid cells. J Bone Miner Res 10 (suppl. 1): S387 (Abstract M539).
31. Diaz R, El-Hajj Fuleihan G, Brown EM, 1998 Regulation of parathyroid function. In: Fray J, ed. Handbook of Physiology, section 7. Endocrinology, Vol. III. Hormonal regulation of water and electrolyte balance. New York: Oxford University Press 607-662.
32. McGehee DS, Aldersberg M, Liu KP, Hsuing S, Heath MJ, Tamir H 1997 Mechanism of extracellular Ca2+ receptor-stimulated hormone release from sheep thyroid parafollicular cells. J Physiol (Lond) 502: 31-44.
33. Hebert SC, Brown EM, Harris HW, 1997 Role of the Ca2+sensing Receptor in divalent mineral ion homeostasis. J Exp Biol 200: 295-302.
34. Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs Jr RW, Lasker RD, 1981 The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Medicine (Baltimore) 60: 397-412.
35. Attie MF, Gill Jr J, Stock JL, Spiegel AM, Downs Jr RW, Levine MA, Marx SJ, 1983 Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest 72: 667-676.
36. Pi M, Garner SC, Flannery P, Spurney RF, Quarles LD, 2000 Sensing of extracellular cations in CasR-deficient osteoblasts. Evidence for a novel cation-sensing mechanism. J Biol Chem 275: 3256-63.
37. Kameda T, Mano H, Yamada Y, Takai H, Amizuka N, Kobori M, Izumi N, Kawashima H, Ozawa H, Ikeda K, Kameda A, Hakeda Y, Kumegawa M, 1998 Calcium-sensing receptor in mature osteoclasts, which are bone resorbing cells. Biochem Biophys Res Commun 245: 419-22.
38. Marx SJ, Attie MF, Stock JL, Spiegel AM, Levine MA, 1981 Maximal urine-concentrating ability: familial hypocalciuric hypercalcemia versus typical primary hyperparathyroidism. J Clin Endocrinol Metab 52: 736-740.
39. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV, 1996 A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 335: 1115-22.
40. Rogers KV, Dunn CK, Hebert SC, Brown EM, 1997 Localization of calcium receptor mRNA in the adult rat central nervous system by in situ hybridization. Brain Res 744: 47-56.
41. Conigrave AD, Quinn SJ, Brown EM, 2000 L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc Natl Acad Sci USA 97: 4814-9.
42. Conigrave AD, Quinn SJ, Brown EM, 2000 Cooperative multi-modal sensing and therapeutic implications of the extracellular Ca(2+) sensing receptor. Trends Pharmacol Sci 21: 401-7.
43. Law Jr WM, Heath III H, 1985 Familial benign hypercalcemia (hypocalciuric hypercalcemia). Clinical and pathogenetic studies in 21 families. Ann Int Med 105: 511-519.
44. Brown EM, 2000 Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin North Am 29: 503-22.
45. Auwerx J, Demedts M, Bouillon R, 1984 Altered para-thyroid set point to calcium in familial hypocalciuric hypercalcaemia. Acta Endocrinologica (Copenh.) 106: 215-218.
46. Khosla S, Ebeling PR, Firek AF, Burritt MM, Kao PC, Heath Hd, 1993 Calcium infusion suggests a “set-point” abnormality of parathyroid gland function in familial benign hypercalcemia and more complex disturbances in primary hyperparathyroidism. J Clin Endocrinol Metab 76: 715-20.
47. Davies M, Adams PH, Lumb GA, Berry JL, Loveridge N, 1984 Familial hypocalciuric hypercalcaemia: evidence for continued enhanced renal tubular reabsorption of calcium following total parathyroidectomy. Acta Endocrinol (Copenh) 106: 499-504.
48. Heath DA, 1994 Familial hypocalciuric hypercalcemia. In: Bilezikian JP, Marcus R, Levine MA, (eds) The Para-thyroids. New York, NY: Raven Press 699-710.
49. Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, Trivedi S, Hellman P, Brown EM, Dahl N, Rastad J, 2000 Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab 85: 2042-7.
50. Spiegel AM, Harrison HE, Marx SJ, Brown EM, Aurbach GD, 1977 Neonatal primary hyperparathyroidism with autosomal dominant inheritance. J Pediatr 90: 269-72.
51. Brown EM, 2000 Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin North Am 29: 503-22.
52. Chou YH, Brown EM, Levi T, Crowe G, Atkinson AB, Arnqvist HJ, Toss G, Fuleihan GE, Seidman JG, Seidman CE, 1992 The gene responsible for familial hypocalciuric hypercalcemia maps to chromosome 3q in four unrelated families. Nat Genet 1: 295-300.
53. Lloyd SE, Pannett AA, Dixon PH, Whyte MP, Thakker RV, 1999 Localization of familial benign hypercalcemia, Oklahoma variant (FBHOk), to chromosome 19q13. Am J Hum Genet 64: 189-95.
54. Heath Hd, Jackson CE, Otterud B, Leppert MF, 1993 Genetic linkage analysis in familial benign (hypocalciuric) hypercalcemia: evidence for locus heterogeneity. Am J Hum Genet 53: 193-200.
55. Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG, 1993 Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75: 1297-303.
56. Bai M, Quinn S, Trivedi S, Kifor O, Pearce SHS, Pollak MR, Krapcho K, Hebert SC, Brown EM, 1996 Expression and characterization of inactivating and activating mutations in the human Ca2+o-sensing receptor. J Biol Chem 271: 19537-45.
57. Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV, 1996 Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest 98: 1860-6.
58. Marx S, Attie M, Spiegel A, Levine M, Lasker R, Fox M, 1982 An association between neonatal severe primary hyperparathyroidism and familial hypocalciuric hypercalcemia in three kindreds. N Engl J Med 306: 257-284.
59. Pollak MR, Chou YH, Marx SJ, Steinmann B, Cole DE, Brandi ML, Papapoulos SE, Menko FH, Hendy GN, Brown EM, et al, 1994 Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest 93: 1108-12.
60. Kobayashi M, Tanaka H, Tsuzuki K, Tsuyuki M, Igaki H, Ichinose Y, Aya K, Nishioka N, Seino Y, 1997 Two novel missense mutations in calcium-sensing receptor gene associated with neonatal severe hyperparathyroidism. J Clin Endocrinol Metab 82: 2716-9.
61. Bai M, Pearce SH, Kifor O, Trivedi S, Stauffer UG, Thakker RV, Brown EM, Steinmann B, 1997 In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2+-sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest 99: 88-96.
62. Bai M, Trivedi S, Brown EM, 1998 Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem 273: 23605-10.
63. Pearce SH, Trump D, Wooding C, Besser GM, Chew SL, Grant DB, Heath DA, Hughes IA, Paterson CR, Whyte MP, et al, 1995 Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest 96: 2683-92.
64. Hosokawa Y, Pollak MR, Brown EM, Arnold A, 1995 Mutational analysis of the extracellular Ca(2+)-sensing receptor gene in human parathyroid tumors. J Clin Endocrinol Metab 80: 3107-10.
65. Chikatsu N, Fukumoto S, Takeuchi Y, Suzawa M, Obara T, Matsumoto T, Fujita T, 2000 Cloning and characterization of two promoters for the human calcium-sensing receptor (CaSR) and changes of CaSR expression in parathyroid adenomas. J Biol Chem 275: 7553-7.
66. Spiegel AM, 1996 Mutations in G protein and G protein-coupled receptors in endocrine disease. J Clin Endocrinol Metab 81: 2434-2442.
67. Parma J, Van Sande J, Swillens S, Tonacchera M, Dumont J, Vassart G, 1995 Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3′,5′-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol 9: 725-33.
68. Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, Hebert SC, Seidman CE, Seidman JG, 1994 Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet 8: 303-7.
69. Baron J, Winer KK, Yanovski JA, Cunningham AW, Laue L, Zimmerman D, Cutler GB, Jr, 1996 Mutations in the Ca(2+)-sensing receptor gene cause autosomal dominant and sporadic hypoparathyroidism. Hum Mol Genet 5: 601-6.
70. Nemeth EF, Bennett SA, 1998 Tricking the parathyroid gland with novel calcimimetic agents [editorial]. Nephrol Dial Transplant 13: 1923-5.
71. Nemeth EF, Fox J, 1999 Calcimimetic Compounds: a Direct Approach to Controlling Plasma Levels of Parathyroid Hormone in Hyperparathyroidism. Trends Endocrinol Metab 10: 66-71.
72. Coburn JW, Maung HM, 2000 Calcimimetic agents and the calcium-sensing receptor. Curr Opin Nephrol Hypertens 9: 123-32.
73. Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, Balandrin MF, 1998 Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA 95: 4040-5.
74. Gowen M, Stroup GB, Dodds RA, James IE, Votta BJ, Smith BR, Bhatnagar PK, Lago AM, Callahan JF, DelMar EG, Miller MA, Nemeth EF, Fox J, 2000 Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest 105: 1595-604.