Abstract
OBJECTIVE: Transsphenoidal (TNS) surgery remains the primary therapeutic option for GH-secreting pituitary adenomas. The aims of this study were to verify the impact of TNS surgery on treatment of acromegaly before and after identification by a dedicated neurosurgical team and to enumerate diagnostic features of the disease described over three decades. DESIGN: 41 patients (group A) who underwent TNS surgery by a dedicated neurosurgical team (2000-2008) and 126 patients (group B) operated on by surgeons not specialized in pituitary surgery (1979-1999) were retrospectively analyzed. RESULTS: No significant differences were observed between the two groups in terms of delay of diagnosis, mean basal GH levels and GH nadir values, prevalence of hypopituitarism and hypertension. IGF-I SDS were significantly higher, while prevalence of IGT/diabetes was significantly lower in group B than in group A. Overall remission rate after surgery was 58.5% for group A (75% in microadenomas and 48% in macroadenomas, P=NS) and 37% for group B (P<0.05 vs group A; for microadenomas, 34% vs 75% of group A, P<0.05, for macroadenomas, 36% vs 48% of group A, P=NS). The mean delay of diagnosis was 4.9 and 5.9 years in group A and B, respectively. CONCLUSIONS: Our data confirm that a dedicated neurosurgical team is needed in order to improve remission rates in acromegalic patients. No changes in biochemical, clinical and neuroradiological presentation of disease were observed over the last three decades. As the high prevalence of macroadenomas negatively influences surgical cure, earlier diagnosis should be considered as mandatory to achieve a better outcome.
INTRODUCTION
Acromegaly is a rare chronic disease caused in almost all patients by a growth hormone-secreting pituitary adenoma, with an annual incidence of 3-4 cases per million and an estimated prevalence of 40-70 cases per million, with an average age at diagnosis of 44 years.
Despite the improvement of biochemical and neuroradiological techniques, the insidious clinical manifestations of acromegaly lead to a delay in diagnosis of from 5 up to 10 years after onset of the disease.1-4 Furthermore, untreated acromegaly is associated with enhanced morbidity and mortality, mainly due to cerebrovascular, cardiovascular, metabolic and respiratory comorbidities, with a 30% decrease in life expectancy. In fact, retrospective studies of acromegalic patients indicate a 10-year reduction in lifespan with at least a doubling of standardized mortality rates (SMRs) due to the detrimental effect of excess of growth hormone (GH) and IGF-I.5,6
Treatment of acromegaly is therefore aimed at reducing GH and IGF-I values to normal range and to correcting symptoms due to the mass effect of the tumor, such as visual defects and headache. According to the Acromegaly Consensus Group current guidelines for disease management, the transsphenoidal (TNS) neurosurgical approach remains a primary option.7 In fact, surgical treatment is able to offer a rapid and, in most cases, permanent reduction in GH and IGF-I levels and to relieve mass effect of the tumor. In those patients in whom surgical treatment is contraindicated or refused, radiotherapy and medical treatment with somatostatin analogs and/or growth hormone receptor antagonists are valuable treatment modalities.
Over recent decades, many data about surgical treatment of GH-secreting pituitary adenomas have been published. However, the remission rates vary widely and are poorly comparable, this owing to heterogeneous criteria used to define biochemical remission of acromegaly (Table 1).8-20 An emerging aspect is the demonstration that the outcome of surgical treatment for acromegaly is strictly influenced by the surgical learning curve and, therefore, by the experience of the neurosurgeon, especially in patients with macroadenoma or extrasellar invasion.11,19-22 Even in the event of a second surgical treatment, a neurosurgical dedicated team is able to improve significantly the final outcome of acromegalic patients.23,24
The aim of this study was to retrospectively investigate the efficacy, tolerability and impact of a dedicated neurosurgical team on the outcome of TNS surgery in 175 acromegalic patients referred to the Neurosurgery Unit of the Fondazione IRCCS Ca’ Granda – Ospedale Maggiore Policlinico of Milan, Italy, between 1979 and 2008. An additional objective was to evaluate over three decades the biochemical and clinical presentation of the disease and the interval from symptoms to diagnosis of acromegaly.
METHODS
Surgical procedure and baseline evaluation
Forty-one acromegalic patients(group A) who underwent neurosurgery for adenomectomy by a dedicated neurosurgical team between 2000 and 2008 at our Institution were investigated. In addition, 126 acromegalic patients (group B) who underwent neurosurgery for adenomectomy by a general neurosurgeon not specialized in pituitary surgery between 1979 and 1999 were also evaluated as a control group. A retrospective database analysis was performed and signed informed consent for data collection was obtained from all patients.
For group A, microscopic TNS surgery with the septal pushover technique was performed.25 For group B, different surgical techniques, such as sublabial or transnasal transsphenoidal approaches, were used.
Starting from 1999, a neuronavigation system (Radionics, Minneapolis, USA; BrainLab, Munich, Germany), loaded with CT and MR coregistered images, was used to verify the surgical anatomy pre- and intraoperatively.
Complications associated with surgical procedure were arbitrarily divided into major (death, carotid lesions, neurological damage, postoperative CSF leak, nasal bleeding or infection) and minor complications (transient diabetes insipidus, nasal discomfort or discharge, headache).
Patients were included in the study if they had clinical and biochemical diagnosis of acromegaly, presence of pituitary adenoma at neuroradiological imaging, no previous radiotherapy or surgical treatment and complete pre- and postoperative follow-up.
The diagnosis of acromegaly was assessed on the basis of clinical signs of acromegaly, failure of suppression of GH to less than 1.0 µg/L during a standard oral glucose tolerance test (OGTT 75 g of glucose) together with serum IGF-I levels greater than the normal age- and sex-adjusted range. Since IGF-I assay was routinely available from 1985, in 26 patients of group B preoperative IGF-I levels were not assessed. OGTT was performed after an overnight fast and collecting blood samples at 0, 30, 60, 90 and 120 minutes. In 25 patients with concomitant diabetes mellitus, samples for serum GH measurement every 30 minutes for 2 hours during slow saline infusion were collected.
Pituitary function was determined via basal or dynamic evaluation, as appropriate. In particular, central hypothyroidism was defined as low FT4 levels in the presence of inappropriate thyroid-stimulating hormone (TSH) levels. Central hypoadrenalism was defined as a lack of response to stimulation tests (peak cortisol levels lower than 500 nmol/l during standard-dose (250 µg) corticotrophin stimulation test (SDCT), low-dose (1 µg) corticotrophin stimulation test (LDCT) or insulin tolerance test). Central hypogonadism was diagnosed in the case of low testosterone or estradiol levels and inappropriately normal or low gonadotropin levels. Prolactin levels were evaluated in all patients.
Moreover, physical examination and metabolic profile (systolic and diastolic blood pressure, glucose and insulin levels at baseline and during OGTT, glycosylated hemoglobin (HbA1c), total cholesterol, high-density lipoprotein (HDL) cholesterol, tryglicerides) were also investigated.
Neuroradiological imaging was performed by magnetic resonance imaging (MRI) or computerized tomography (CT) scan. According to size, pituitary tumors were classified into microadenomas (maximum diameter <10 mm) or macroadenomas (maximum diameter equal or greater than 10 mm). In 4 out of 126 patients of group B, classification was not performed because of available imaging (X-ray).
Postoperative evaluation
Hormonal and metabolic assessment was performed in all patients in the postoperative period (short-term evaluation) with a mean distance from TNS procedure of 2.9±1.3 months (range: 2-10 months). In the event of preoperative medical therapy, evaluation was performed at least 3 months after surgery.
At short-term evaluation, patients were considered surgically cured in the presence of normal serum IGF-I levels and a GH nadir <1 µg/L after OGTT. Similarly to baseline evaluation, in 23 patients of group B IGF-I assay was not available. In these patients, we considered GH nadir <1 µg/L as the criteria to define surgical cure. Patients with discrepant biochemical results (normal GH levels and elevated IGF-I levels, as well as elevated GH levels and normal IGF-I values) were considered not cured.
Assay methods
Until 1992, serum GH levels were measured via radioimmunoassay (RIA) with the polyclonal Lisophase HGH kit (Sclavo, Milan, Italy). The sensitivity of the method was 0.3 mg/L . The standards were calibrated against the first World Health Organization International Reference Preparation HGH-MCR (code 66/217). Subsequently and prior to October 2007, a two-site monoclonal immunofluorimetric assay method (AutoDelfia kit, Wallac, Inc. OY, Turku, Finland) was used. The sensitivity of this method was 0.01 mg/L and intra- and interassay coefficients of variation were 2% and 1.7%, respectively. After October 2007, GH was assayed with a chemiluminescence method (Immulite 2000, Siemens Medical Solutions Diagnostics, Los Angeles, CA) with a detection limit of 0.01 mg/l. In both cases, the standards were calibrated with the first World Health Organization International Reference Preparation (code 80/505).
Serum IGF-I concentrations were measured by commercial RIA kits starting from 1985. According to the RIA assay used before 1996 (Incstar, Stillwater, MN, USA), the removal of binding proteins was obtained by acidification and subsequent filtration on ODS C18 cartridges. The intra- and interassay coefficients of variation were 15 and 16%, respectively. Afterwards and until 2008, IGF-I levels were assessed by the commercial radioimmunometric assay kit of Mediagnost (Tübingen, Germany). The separation of IGF-I from binding proteins was obtained by acidification in IGF-II excess, and IGF-II cross-reactivity was less than 0.05%. The intra- and interassay coefficients of variation were 3.2 and 8.9%, respectively. The values were compared with those from an appropriate age- and sex-adjusted range and expressed in standard deviation scores (SDS).
All the other hormonal, biochemical and metabolic parameters were measured by standard procedures.
Statistical analysis
Calculations were performed by SPSS 18.0 software (SPSS, Paris, France). Data are presented as mean ± standard deviation (SD). Normally distributed variables were compared using Student’s t-test, preceded by Levene’s test to check variance equality. Nominal data were analyzed by Fisher’s exact test in 2 × 2 contingency tables or χ2 test otherwise, while the other non-gaussian variables were evaluated by the Mann-Whitney U-Test. Logistic multivariate regression analysis was performed with the stepwise method. P <0.05 and P >0.10 were used as entry and removal criteria, respectively. Two-tailed P values less than 0.05 were considered statistically significant.
RESULTS
Baseline evaluation
In group A (n=41, 19F & 22M, mean age at diagnosis 47.3±13.3 years), the mean duration of symptoms before diagnosis of acromegaly was 4.9±3.6 years. Sixteen patients (39%) had a microadenoma, while the remaining 25 (61%) had a macroadenoma. Mean basal GH values and mean nadir of GH after OGTT were 23.9±29.7 µg/L and 19.8±32.9 µg/L, respectively, whereas IGF-I SDS were 10±5.5. Mean age at the surgical procedure was 48.2±13.6 yrs. At diagnosis, single or multiple pituitary deficits were diagnosed in 16 out of 41 patients (39%). In particular, 12 patients had one pituitary deficit (9 hypogonadism, 2 hypoadrenalism, 1 hypothyroidism), while multiple deficiencies were observed in 4 cases (3 hypogonadism and hypoadrenalism, 1 hypogonadism and hypothyroidism). In 7 patients, high prolactin levels, indicating mixed lacto/somatotroph tumors or deafferentation, were observed.
Headache was present in 22 patients (54%). Visual field examination, performed in all the 25 patients with macroadenomas, showed a defect in 5 cases (12%). Before surgery, 13 patients (32%) received medical therapy (10 somatostatin analogs, 2 somatostatin analogs and dopamine agonists, 1 somatostatin analogs and pegvisomant).
In group B (n=126, 79F & 47M, mean age at diagnosis 43.3±10.7 years), the mean duration of symptoms before diagnosis of acromegaly was 5.9±5 years. There were 35 of our cases were having microadenomas (i.e. 29% of our population) while 87 macroadenomas (71%). In 4 patients tumor extension was undetermined. Mean basal GH and mean GH nadir levels were 28.8±28.1 µg/L and 19.9±20.9 µg/L, respectively, IGF-I SDS were 15.4±6.4. Mean age at the surgical procedure was 44.1±10.6 yrs. Single or multiple pituitary deficits were present in 31/126 patients (25%). In particular, 22 patients had one pituitary deficit (19 hypogonadism, 2 hypoadrenalism, 1 hypothyroidism), while multiple deficiencies were observed in 9 cases (7 hypogonadism and hypothyroidism, 1 hypogonadism and hypoadrenalism, 1 hypoadrenalism, hypogonadism and hypothyroidism). Elevated prolactin levels were found in 26 patients (21%). Headache was present in 78 patients (62%). Before surgery, 14 patients (11%) received medical therapy (11 somatostatin analogs, 3 dopamine agonists).
No significant differences between group A and group B in terms of delay of diagnosis, mean age, tumor size, mean basal GH levels, mean GH nadir after OGTT, prevalence of hypopituitarism and hypertension were observed. IGF-I SDS were significantly higher in group B (15.4±6.4 vs 10±5.5, P<0.001). Finally, prevalence of diabetes mellitus or impaired glucose tolerance (IGT) were higher in group A than in group B.
Demographic, neuroradiological, biochemical and clinical characteristics of the two groups of patients are summarized in Table 2.
Transsphenoidal surgery
No case of deaths related to surgery was recorded. Morbidity was very low: specifically, as far as major complications are concerned, postoperative cerebrospinal fluid leak was reported in 1 patient (3%) of group A, treated with intravenous antibiotics, and in 5 patients (4%) of group B, successfully treated with lumbar drainage in 4 cases and intravenous antibiotics in 1 case. Regarding minor complications, transient diabetes insipidus was observed in 4 patients (10%) of group A and 8 patients (6%) of group B, spontaneously resolved in all patients before the discharge.
Postoperative evaluation
At first evaluation after surgery (mean distance from TNS: 2.9±1.3 months, range 2-10 months), disease remission was observed in 24 out of 41 patients (58.5%) of group A. In particular, surgical cure was observed in 12/16 patients (75%) with microadenomas and in 12/25 patients (48%) with macroadenomas. No significant difference in remission rates between micro- and macroadenomas was seen.
As expected, mean GH levels, mean GH nadir after OGTT and IGF-I SDS were significantly lower with respect to presurgical evaluation (23.9±29.7 vs 3.5±4.3 µg/L, 19.8±32.9 vs 2.7±4.7 µg/L and 10±5.5 vs 2.9±5.4, respectively, for all P<0.001). Furthermore, a significant reduction of the prevalence of headache was observed after TNS (from 55% to 20%, P=0.002).
Concerning pituitary function after surgery, in group A single or multiple pituitary deficits were diagnosed in 20 patients (49%). Notably, 10/13 patients with preoperative diagnosis of hypogonadism gained normal function of the pituitary/gonadal axis after surgery. By contrast, new diagnosis of hypoadrenalism, hypogonadism and hypothyroidism was recorded in 12, 3 and 6 patients, respectively.
Finally, no difference in remission rates between patients receiving or not receiving preoperative medical therapy [61% (8/13) vs 57% (16/28), P=1.000] was observed.
In group B, surgical cure was achieved in 47 out of 126 patients (37% vs 58.5% of group A, P=0.019). With regard to tumor size, disease remission was observed in 12/35 patients of microadenomas (34% vs 75% of group A, P=0.014) and in 31/87 patients with macroadenomas (36% vs 48% of group A, P=0.350) (Figure 1). Mean GH levels, mean GH nadir after OGTT and IGF-I SDS were significantly lower than those of the presurgical evaluation (28.8±28.1 vs 8.1±14.6 µg/L, 19.9±20.9 vs 4.5±8.8 µg/L, and 15.4±6.4 vs 5.5±6.4, respectively, for all P<0.001). However, postoperatively, mean GH values and IGF-I SDS were significantly higher in patients of group B compared with those of group A (8.1±14.6 vs 3.5±4.3 µg/L and 5.5±6.4 vs 2.9±5.4, respectively, P=0.048 and P=0.020). Similarly to group A, after TNS a significant reduction of the prevalence of headache was observed (from 61% to 28%, P<0.001).
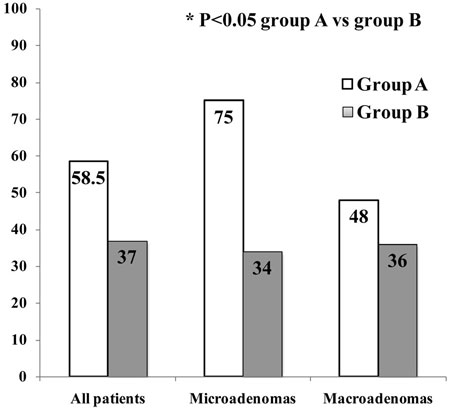
Figure 1. Prevalence of remission rates after TNS surgery in two groups of acromegalic patients.
Single or multiple pituitary deficits were diagnosed in 32 patients (25.4% vs 49% of group A, P=0.006). In 22/28 patients with hypogonadism, in 2/3 with hypoadrenalism and in 6/10 with hypothyroidism prior surgery, normal function of the respective axis after TNS was observed. A new diagnosis of hypoadrenalism, hypogonadism and hypothyroidism was performed in 17, 6 and 7 patients, respectively.
Finally, in order to determine the factors that affect surgical cure in acromegalic patients, a stepwise multivariate logistic regression analysis was performed. In this regression model, sex, age at diagnosis, basal GH levels, basal IGF-I levels, belonging to group A or group B and tumor size were included. Among all the independent variables, basal GH levels [OR: 0.961 (0.935-0.987), P=0.004], basal IGF-I levels [OR: 0.985 (0.974-0.997), P=0.015] and belonging to group A or Group B [OR: 2.618 (1.028-6.670), P 0.044] were the significant determinants of surgical cure.
DISCUSSION
Acromegaly is an insidious disease associated with enhanced morbidity and mortality, with a 30% decrease in life expectancy. The aims of acromegaly treatment are normalization of GH and IGF-I values and control of tumor growth. Transsphenoidal surgery is the primary therapy for GH-secreting pituitary adenomas, in particular for intrasellar microadenomas, noninvasive macroadenomas and in the presence of compression symptoms.7
To date, many studies focusing on remission rates of surgically treated GH-secreting pituitary adenomas have been published. Although the results are poorly comparable because of different criteria used to define biochemical cure, overall surgical remission rates vary from 54.3 to 82%.8-20
Several authors have previously observed that, although tumor volume and invasiveness are factors influencing the success of surgical procedure,24 outcome of TNS surgery is strictly dependent on the availability of a dedicated neurosurgical team.11,19-22
In our series, neuroradiological imaging showed the presence of a macroadenoma in the majority of patients (61% in group A and 71% in group B). After surgery, we observed a significant increase in overall remission rate in patients surgically treated by a dedicated neurosurgical team (58.5%) in respect to patients who underwent TNS surgery by a non-selected operator (37%).
With regard to the tumor size within groups, we did not observe a significant difference in remission rate between patients affected by microadenoma and patients affected by macrodenoma in both groups A and B. This result is not in agreement with some surgical series that report a better outcome for microadenomas: this may be related, at least for group A, to the relatively small numbers of patients analyzed.12,14,19
Furthermore, a significant increase in surgical cure was observed in patients of group A with microadenomas, but not in patients with macroadenomas, as compared to patients belonging to group B. Even though remission rate of macroadenomas did not increase between the two groups, surgical results of group A are comparable to those already published in the literature.8-20 Moreover, it has been shown that surgical outcome improves with time and consequently with neurosurgical experience.12 Thus, TNS treatment performed by our neurosurgical team requires further and later evaluation.
Notably, stepwise regression analysis of our series showed that basal GH, IGF-I levels and belonging to group A or B, but not tumor size, were the determinants of surgical cure. This observation, consistent with the similar outcome that we observed between micro- and macroadenomas, may reflect the poor characterization of macroadenomas in terms of local invasiveness, which represents a limitation of this retrospective work.
The influence of preoperative medical therapy, in particular with somatostatin analogs, on surgical outcome is controversial.7 Relative to previous data, in our patients of group A remission rate after surgery did not improve in the event of preoperative medical treatment.16
Regarding complications of TNS surgery, our series showed absent mortality and very low morbidity. As far as pituitary function is concerned, data about pre- and postoperative pituitary insufficiency in the literature are inconclusive.16,19 In our study, a similar percentage of improvement or worsening of pituitary secretion after surgery was observed in two groups of patients. However, while gonadal function seemed to often resolve after surgical treatment, a new diagnosis of hypoadrenalism and hypothyroidism was frequently performed. Although the reason for this circumstance is unclear, our results confirm that a complete pre- and postoperative pituitary function evaluation is necessary.
We did not observe significant changes in clinical, biochemical and neuroradiological presentation of acromegaly over the last three decades. It is well known that acromegaly is a rare and insidious disease the diagnosis of which is often late. Older series reported a mean distance of diagnosis from onset of symptoms of 5 to 10 years.1-3,26-28 Although Nachtigall and coll. showed a mean time to diagnosis of 2.5 years,29 more recently Reid and coll. confirmed a mean delay of more than 5 years in a large series of consecutive acromegalic patients diagnosed between 1981 and 2006.4 Similarly, our results showed a mean duration of symptoms before diagnosis of 5.5 and 5.9 years in group A and in group B, respectively. In agreement with the similar time to diagnosis in groups A and B, tumor size, mean basal GH levels, mean GH nadir after OGTT and prevalence of hypopituitarism and hypertension did not differ between groups. Conversely, prevalence of diabetes mellitus or IGT was higher in group A than in group B, this result probably reflecting a higher prevalence of preoperative treatment with somatostatin analogs in group A.
In conclusion,our data confirm that a dedicated neurosurgical team is needed in order to improve remission rates of surgery in acromegalic patients, in particular for patients with microadenomas. Moreover, we did not observe significant changes in biochemical, clinical and neuroradiological presentation of the disease over the last three decades. Therefore, despite the improvement of biochemical, neuroradiological and neurosurgical techniques, acromegaly remains under-diagnosed. The high proportion of macroadenomas at diagnosis is an important challenge: in this context, surgery is not curative in a significant proportion of cases and achievement of disease control requires medical therapy and/or radiotherapy. Hence, as the high prevalence of macroadenomas negatively influences surgical cure, a strategy enabling enhanced awareness and an earlier diagnosis of acromegaly should be considered as mandatory to achieve an even better outcome.
FUNDING
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
COMPETING INTEREST
None.
REFERENCES
1. Ezzat S, Forster MJ, Berchtold P, Redelmeier D, 1994 Acromegaly: clinical and biochemical features in 500 patients. Medicine 73: 233-240.
2. Holdaway IM, Rajasoorya RC, 1999 Epidemiology of acromegaly. Pituitary 2: 29-41.
3. Holdaway IM, Rajasoorya RC, Gamble GD, 2004 Factors influencing mortality in acromegaly. J Clin Endocrinol Metab 89: 667-674.
4. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU, 2010 Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf) 72: 203-208.
5. Holdaway IM, Bolland MJ, Gamble GD, 2008 A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol 159: 89-95.
6. Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP, 2008 Mortality in Acromegaly: A Metaanalysis. J Clin Endocrinol Metab 93: 61-67.
7. Melmed S, Colao A, Barkan A, et al, 2009 Guidelines for acromegaly management: an update. J Clin Endocrinol Metab 94: 1509-1517.
8. Losa M, Oeckler R, Schopohl J, Müller OA, Alba-Lopez J, von Werder K, 1989 Evaluation of selective transsphenoidal adenomectomy by endocrinological testing and somatomedin-C measurement in acromegaly. J Neurosurg 70: 561-567.
9. Tindall GT, Oyesiku NM, Watts NB, Clark RV, Christy JH, Adams DA, 1993 Transsphenoidal adenomectomy for growth hormone-secreting pituitary adenomas in acromegaly: outcome analysis and determinants of failure. J Neurosurg 78: 205-215.
10. Fahlbusch R, Honegger J, Buchfelder M, 1997 Evidence supporting surgery as treatment of choice for acromegaly. J Endocrinol 155: Suppl 1: 53-55.
11. Lissett CA, Peacey SR, Laing I, Tetlow L, Davis JR, Shalet SM, 1998 The outcome of surgery for acromegaly: the need for a specialist pituitary surgeon for all types of growth hormone (GH) secreting adenoma. Clin Endocrinol (Oxf) 49: 653-657.
12. Ahmed S, Elsheikh M, Stratton IM, Page RC, Adams CB, Wass JA, 1999 Outcome of transsphenoidal surgery for acromegaly and its relationship to surgical experience. Clin Endocrinol (Oxf) 50: 561-567.
13. Shimon I, Cohen ZR, Ram Z, Hadani M, 2001 Transsphenoidal surgery for acromegaly: endocrinological follow-up of 98 patients. Neurosurgery 48: 1239-1243.
14. Nomikos P, Buchfelder M, Fahlbusch R, 2005 The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol 152: 379-387.
15. Abbassioun K, Amirjamshidi M, Mehrazin A, et al, 2006 A prospective analysis of 151 cases of patients with acromegaly operated by one neurosurgeon: a follow-up of more than 23 years. Surg Neurol 66: 26-31.
16. Petersenn S, Buchfelder M, Reincke M, et al, 2008 Results of surgical and somatostatin analog therapies and their combination in acromegaly: a retrospective analysis of the German Acromegaly Register. Eur J Endocrinol 159: 525-532.
17. Gondim JA, Almeida JP, de Albuquerque LA, Gomes E, Schops M, Ferraz T, 2010 Pure endoscopic transsphenoidal surgery for treatment of acromegaly: results of 67 cases treated in a pituitary center. Neurosurg Focus 29: E7.
18. Kristof RA, Grote A, Redel L, Neuloh G, Klingmüller D, Schramm J, 2011 The common consensus criteria have high predictive values for long-term postoperative acromegaly remission. Acta Neurochir (Wien) 153: 19-25.
19. Dusek T, Kastelan D, Melada A, et al, 2011 Clinical features and therapeutic outcomes of patients with acromegaly: single-center experience. J Endocrinol Invest 34: e382-385.
20. Wang M, Mou C, Jiang M, et al, 2012 The characteristics of acromegalic patients with hyperprolactinemia and the differences in patients with merely GH-secreting adenomas: clinical analysis of 279 cases. Eur J Endocrinol 166: 797-802.
21. Gittoes NJ, Sheppard MC, Johnson AP, Stewart PM, 1999 Outcome of surgery for acromegaly—the experience of a dedicated pituitary surgeon. QJM 92: 741-745.
22. Leach P, Abou-Zeid AH, Kearney T, Davis J, Trainer PJ, Gnanalingham KK, 2010 Endoscopic transsphenoidal pituitary surgery: evidence of an operative learning curve. Neurosurgery 67: 1205-1212.
23. Boeving A, Borba LA, Rodrigues AM, et al, 2006 Outcome of surgical treatment for acromegaly performed by a single neurosurgeon and cumulative meta analysis. Endocrinol Metabol 50: 884-892.
24. Ludecke DK, Abe T, 2006 Transsphenoidal microsurgery for newly diagnosed acromegaly: a personal view after more 1,000 operations. Neuroendocrinology 83: 230-239.
25. Powell M, 2012 Microscope transsphenoidal surgery. Acta Neurochir (Wien) 154: 913-917.
26. Alexander L, Appleton D, Hall R, Ross WM, Wilkinson R, 1980 Epidemiology of acromegaly in the Newcastle region. Clin Endocrinol (Oxf) 12: 717-719.
27. Nabarro JD, 1987 Acromegaly. Clin Endocrinol (Oxf) 26: 481-512.
28. Bengtsson BA, Edén S, Ernest I, Odén A, Sjögren B, 1988 Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984. Acta Med Scand 223: 327-335.
29. Nachtigall L, Delgado A, Swearingen B, Lee H, Zerikly R, Klibanski A, 2008 Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab 93: 2035-2041.
Address for correspondence:
Dr. Emanuele Ferrante, Endocrinology and Diabetology Unit – Pad. Granelli, Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Via F. Sforza, 35, 20122 Milan, Italy, Tel. +39 02 50320608; Fax +39 02 50320605, E-mail: leleferrante@gmail.com
Received 15-07-2013, Accepted 18-12-2013